From Surf Wiki (app.surf) — the open knowledge base
TAR DNA-binding protein 43
Protein found in humans
Protein found in humans
Transactive response DNA binding protein 43 kDa (TAR DNA-binding protein 43 or TDP-43) is a protein that in humans is encoded by the TARDBP gene.
Structure
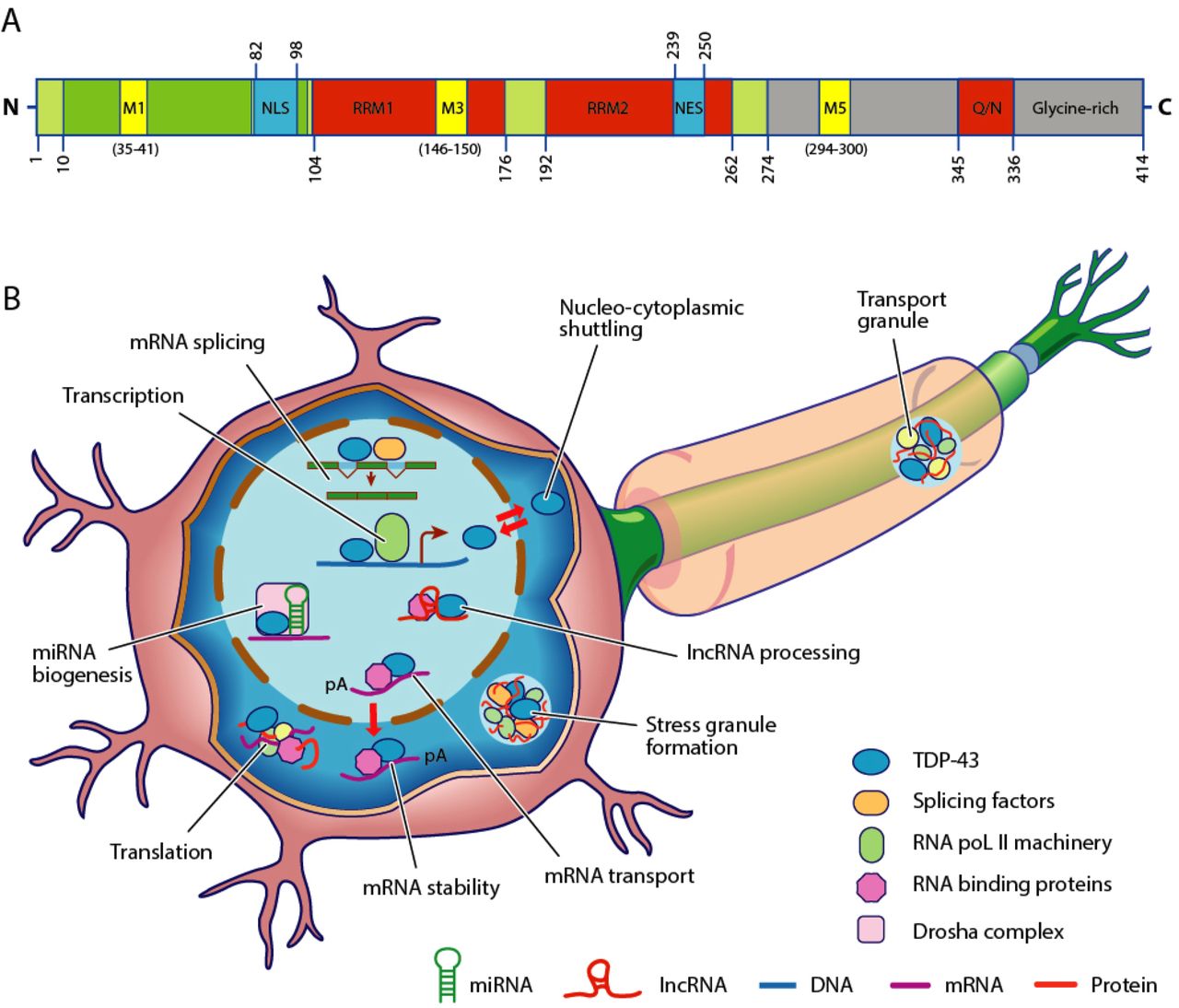
TDP-43 is 414 amino acid residues long. It consists of four domains: an N-terminal domain spanning residues 1–76 (NTD) with a well-defined fold that has been shown to form a dimer or oligomer; two highly conserved folded RNA recognition motifs spanning residues 106–176 (RRM1) and 191–259 (RRM2), respectively, required to bind target RNA and DNA; an unstructured C-terminal domain encompassing residues 274–414 (CTD), which contains a glycine-rich region, is involved in protein-protein interactions, and harbors most of the mutations associated with familial amyotrophic lateral sclerosis.
The entire protein devoid of large solubilising tags has been purified. The full-length protein is a dimer. The dimer is formed due to a self-interaction between two NTD domains, where the dimerisation can be propagated to form higher-order oligomers.
The protein sequence also has a nuclear localization signal (NLS, residues 82–98), a former nuclear export signal (NES residues 239–250) and 3 putative caspase-3 cleavage sites (residues 13, 89, 219).
In December 2021 the structure of TDP-43 was resolved with cryo-EM but shortly after it was argued that in the context of FTLD-TDP the protein involved could be TMEM106B (which has been also resolved with cryo-EM), rather than of TDP-43.
N-Terminal domain (NTD)
The NTD located between residues 1 and 76 is involved in TDP-43 polymerization. Indeed, dimers are formed by head-to-head interactions between NTDs, and the polymer thus obtained allows for pre-mRNA splicing. However, further oligomerization brings to more toxic accumulates. This process of polymerization into dimers, larger forms or just stabilizing monomers is dependent on TDP-43 conformational equilibrium between monomers, homodimers and oligomers. Hence, in TDP-43 diseased cells, TDP-43's over-expression leads to the NTD showing high propensity to aggregate. Contrary to this, in normal cells, normal levels of TDP-43 allow for folded NTD, preventing aggregates and polymers formation.
More recently, this domain was found to have a ubiquitin-like structure. It bears 27,6% of homology with Ubiquitin-1 and a β1-β2-α1-β3-β4-β5-β6 + 2*SO42- form. Ubiquitin-like domain are usually associated with a greater affinity for RNA/DNA. However, in the unique case of TDP-43, the Ubiquitin-like NTD binds directly to ssDNA. This interaction permits the conformational equilibrium cited higher to shift towards non-aggregated forms.
The domain spanning from [1,80] has a solenoid-like structure which sterically impedes interactions between aggregation prone C-term regions.
All of this raises the possibility that NTD and the RNA recognition motifs (later on defined) could cooperatively interact with nucleic acids to accomplish TDP-43's physiological functions.
Mitochondrial localization signal
There are six mitochondrial localization signals to be accounted on TDP-43's amino acid sequence, although only M1, M3, and M5 were shown to be essential for mitochondrial localization. Indeed, their ablation leads to a lessened mitochondrial localization.
These localizing sequences are found on the following amino acids:
M1: [35, 41], M2: [105, 112], M3: [146-150], M4: [228, 235], M5: [294, 300], M6: [228, 236].
Nuclear localization signal (NLS)
The nuclear localization signal (NLS) domain is located between residues 82 and 98 is of critical importance in ALS, and such is witnessed by the depletion or the mutations (notably A90V) of this domain, which cause loss-of-function from nucleus and promote aggregating, two processes very likely to conduct to TDP-43's toxic gain of function.
It is thereby of the utmost importance to note that TDP-43's nuclear localization is absolutely critical for it to fulfill its physiological functions.
RNA recognition motif
The RNA recognition motif ranges between residues 105 and 181, much like many hnRNPs, TDP-43's RRMs encompass highly conserved motifs of primary importance for fulfilling their function. Both RRMs follow this pattern: β1-α1-β2-β3-α2-β4-β5, which allows them to bind to both RNA and DNA onto U G/T G-repeats of 3'UTR (Untranslated Terminal Regions) end of mRNA/DNA.
These sequences mainly ensure mRNA processing, RNA export and RNA stabilizing. It is notably thanks to these sequences that TDP-43 importantly binds to its own mRNA regulates its very own solubility and polymerization.
RRM2
RRM2 spans between residues 181 and 261. In pathological conditions, it notably binds to p65/NF-kB, an apoptosis implicated factor, and is thus a potential therapeutic target. Moreover it can be burdened with a mutation, D169G, altering a key cleaving site for regulating formation of toxic inclusions.
Nuclear export signal (NES)
The nuclear export signal is located between residues 239 and 251 sequence probably bears a role in TDP-43's shuttling function, and was recently found using a prediction algorithm.
Disordered glycin rich C-terminal domain (CTD)
The Disordered Glycin Rich C-terminal domain is located between residues 277 and 414. Much like 70 other RNA binding proteins, TDP-43 bears a Q/N rich domain [344, 366] which resembles yeast prion sequence. This sequence is called a Prion-Like Domain (PLD).
PLDs are low complexity sequences that have been reported to mediate gene regulation via Liquid-Liquid Phase Transition (LLP) thus driving RNP granule assembly. Forming these microscopically visible RNP granules is thought to induce more effective gene regulatory process.
It is here noted that LLP are reversible phenomenons of de-mixing a solution into two distinct liquid phases, hereby forming granules.
Mutations within the TDP-43 proteins Glycine Rich Region (GRR) have recently been identified as associates that can contribute to various neurodegenerative diseases, with the most notable and common NDD being ALS, about 10% of the mutations causing familial ALS are accredited with the TDP-43 protein
This CTD is often reported to play important role in pathogenic behavior of TDP-43:
RNPs granules could have a role in stress response, and thus, aging, or persistence stress could lead the LLPs to turn into irreversible Liquid Solid Phase separation, pathological aggregates notably found in ALS neurons.
The CTD's disorganized structure can turn into a full fledged amyloid-like beta-sheet rich structure, causing it to adopt prion-like properties.
Moreover, CTFs are a common marker in diseased neurons and are argued to be of high toxicity.
However, notice is to be taken that some points are not always consensual. Indeed, due to its hydrophobic structure, TDP-43 can be hard to analyze, and parts of it remain somewhat vague. Precise sites of phosphorylation, methylation, or even binding are still a bit elusive.
Function
TDP-43 is a transcriptional repressor that binds to chromosomally integrated TAR DNA and represses HIV-1 transcription. In addition, this protein regulates alternate splicing of the CFTR gene. In particular, TDP-43 is a splicing factor binding to the intron8/exon9 junction of the CFTR gene and to the intron2/exon3 region of the apoA-II gene. A similar pseudogene is present on chromosome 20.
TDP-43 has been shown to bind both DNA and RNA and have multiple functions in transcriptional repression, pre-mRNA splicing and translational regulation. Recent work has characterized the transcriptome-wide binding sites revealing that thousands of RNAs are bound by TDP-43 in neurons.
TDP-43 was originally identified as a transcriptional repressor that binds to chromosomally integrated trans-activation response element (TAR) DNA and represses HIV-1 transcription. It was also reported to regulate alternate splicing of the CFTR gene and the apoA-II gene.
In spinal motor neurons TDP-43 has also been shown in humans to be a low molecular weight neurofilament (hNFL) mRNA-binding protein. It has also shown to be a neuronal activity response factor in the dendrites of hippocampal neurons suggesting possible roles in regulating mRNA stability, transport and local translation in neurons.
It has been demonstrated that zinc ions are able to induce aggregation of endogenous TDP-43 in cells. Moreover, zinc could bind to RNA binding domain of TDP-43 and induce the formation of amyloid-like aggregates in vitro. Consistently, Zn2+ binds a short peptide (residues 256–264) from the C-terminus of TDP-43's RRM2 domain with an association constant of ~1.6×105 M−1, supporting a specific Zn2+ site that may regulate nucleic-acid binding.
DNA repair
TDP-43 protein is a key element of the non-homologous end joining (NHEJ) enzymatic pathway that repairs DNA double-strand breaks (DSBs) in pluripotent stem cell-derived motor neurons. TDP-43 is rapidly recruited to DSBs where it acts as a scaffold for the further recruitment of the XRCC4-DNA ligase protein complex that then acts to seal the DNA breaks. In TDP-43 depleted human neural stem cell-derived motor neurons, as well as in sporadic ALS patients' spinal cord specimens there is significant DSB accumulation and reduced levels of NHEJ.
Clinical significance
A hyper-phosphorylated, ubiquitinated and cleaved form of TDP-43—known as pathologic TDP43—is the major disease protein in ubiquitin-positive, tau-, and alpha-synuclein-negative frontotemporal dementia (FTLD-TDP, previously referred to as FTLD-U) and in amyotrophic lateral sclerosis (ALS). Elevated levels of the TDP-43 protein have also been identified in individuals diagnosed with chronic traumatic encephalopathy, and has also been associated with ALS leading to the inference that athletes who have experienced multiple concussions and other types of head injury are at an increased risk for both encephalopathy and motor neuron disease (ALS). Abnormalities of TDP-43 also occur in an important subset of Alzheimer's disease patients, correlating with clinical and neuropathologic features indexes. Misfolded TDP-43 is found in the brains of older adults over age 85 with limbic-predominant age-related TDP-43 encephalopathy, (LATE), a form of dementia. New monoclonal antibodies, 2G11 and 2H1, have been developed to specify different TDP-43 inclusion types that occur across neurodegenerative diseases, without relying on hyper-phosphorylated epitopes. These antibodies were raised against an epitope within the RRM2 domain (amino acid residues 198–216).
Mutations in the TARDBP gene are associated with neurodegenerative disorders including frontotemporal lobar degeneration and amyotrophic lateral sclerosis (ALS). In particular, the TDP-43 mutants M337V and Q331K are being studied for their roles in ALS. While the aberrant mislocalization and cytoplasmic aggregation of TDP-43 characterizes FTLD with TDP-43 pathology (FTLD-TDP), recent work suggests the amyloid fibrils found in human FTLD-TDP brains are composed of transmembrane lysosomal protein TMEM106b rather than TDP-43. Cytoplasmic TDP-43 pathology is the dominant histopathological feature of multisystem proteinopathy. The N-terminal domain, which contributes importantly to the aggregation of the C-terminal region, has a novel structure with two negatively charged loops. A recent study has demonstrated that cellular stress can trigger the abnormal cytoplasmic mislocalisation of TDP-43 in spinal motor neurons in vivo, providing insight into how TDP-43 pathology may develop in sporadic ALS patients.
Figures
References
References
- (June 1995). "Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs". Journal of Virology.
- (June 2017). "Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation". Nature Communications.
- (March 2018). "A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing". The EMBO Journal.
- (March 1986). "Role of calcium in the phloretin effects on sugar transport in rat small intestine". Revista Espanola de Fisiologia.
- (September 2016). "ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain". Structure.
- (October 2019). "Isolation and characterization of soluble human full-length TDP-43 associated with neurodegeneration". FASEB Journal.
- (January 2022). "Structure of pathological TDP-43 filaments from ALS with FTLD". Nature.
- "An ALS Protein, Revealed".
- (March 2022). "Amyloid fibrils in disease FTLD-TDP are composed of TMEM106B not TDP-43". Nature.
- "Frontotemporal Dementia: Not the Protein We Thought".
- (December 2014). "TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA". Proceedings of the National Academy of Sciences of the United States of America.
- (2019-02-14). "Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis". Frontiers in Molecular Neuroscience.
- "TARDBP TAR DNA binding protein [Homo sapiens (human)] - Gene - NCBI".
- (December 2014). "TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA". Proceedings of the National Academy of Sciences of the United States of America.
- (August 2016). "Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins". Journal of Neurochemistry.
- (October 2020). "Maintaining the balance of TDP-43, mitochondria, and autophagy: a promising therapeutic strategy for neurodegenerative diseases". Translational Neurodegeneration.
- (February 2019). "Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology". The Journal of Clinical Investigation.
- (June 2007). "TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein". Molecular and Cellular Neurosciences.
- (July 2013). "Prion-like properties of pathological TDP-43 aggregates from diseased brains". Cell Reports.
- (2016). "RNA Processing". Springer International Publishing.
- (August 2020). "The role of TDP-43 mislocalization in amyotrophic lateral sclerosis". Molecular Neurodegeneration.
- (August 2015). "Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles". The Journal of Cell Biology.
- (April 2001). "Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping". The EMBO Journal.
- (April 2009). "Structural insights into TDP-43 in nucleic-acid binding and domain interactions". Nucleic Acids Research.
- [https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=ShowDetailView&TermToSearch=23435 Gene Result]
- (January 2011). "Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes". The Journal of Biological Chemistry.
- (September 2001). "Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9". The Journal of Biological Chemistry.
- (May 2008). "TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor". Journal of Neurochemistry.
- (May 2010). "Zinc induces depletion and aggregation of endogenous TDP-43". Free Radical Biology & Medicine.
- (July 2017). "Zinc binding to RNA recognition motif of TDP-43 induces the formation of amyloid-like aggregates". Scientific Reports.
- (2020). "Zinc Binds to RRM2 Peptide of TDP-43". International Journal of Molecular Sciences.
- (March 2019). "Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects". Proceedings of the National Academy of Sciences of the United States of America.
- (July 2011). "A harmonized classification system for FTLD-TDP pathology". Acta Neuropathologica.
- (April 2018). "Prion-like properties of disease-relevant proteins in amyotrophic lateral sclerosis". Journal of Neural Transmission.
- (February 2018). "Disruption of ER-mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis". Cell Death & Disease.
- [[Alan Schwarz. Schwarz, Alan]]. [https://www.nytimes.com/2010/08/18/sports/18gehrig.html "Study Says Brain Trauma Can Mimic A.L.S."], ''[[The New York Times]]'', August 18, 2010. Accessed August 18, 2010.
- (September 2011). "Accumulation of transactive response DNA binding protein 43 in mild cognitive impairment and Alzheimer disease". Journal of Neuropathology and Experimental Neurology.
- (November 2020). "Novel monoclonal antibodies targeting the RRM2 domain of human TDP-43 protein". Neuroscience Letters.
- (July 2007). "TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease". Acta Neuropathologica.
- (March 2008). "TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis". Science.
- (2013). "TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43". Journal of Alzheimer's Disease.
- (2019). "Molecular Mechanisms of Neurodegeneration Related to ''C9orf72'' Hexanucleotide Repeat Expansion". Behavioural Neurology.
- (May 2022). "Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43". Nature.
- (March 2013). "Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS". Nature.
- (April 2016). "The TDP-43 N-terminal domain structure at high resolution". The FEBS Journal.
- (September 2018). "Nucleo-cytoplasmic transport of TDP-43 studied in real time: impaired microglia function leads to axonal spreading of TDP-43 in degenerating motor neurons". Acta Neuropathologica.
- (November 2020). "TDP-43 proteinopathies: a new wave of neurodegenerative diseases". Journal of Neurology, Neurosurgery, and Psychiatry.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about TAR DNA-binding protein 43 — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report