From Surf Wiki (app.surf) — the open knowledge base
Missense mutation
Genetic point mutation that results in an amino acid change in a protein
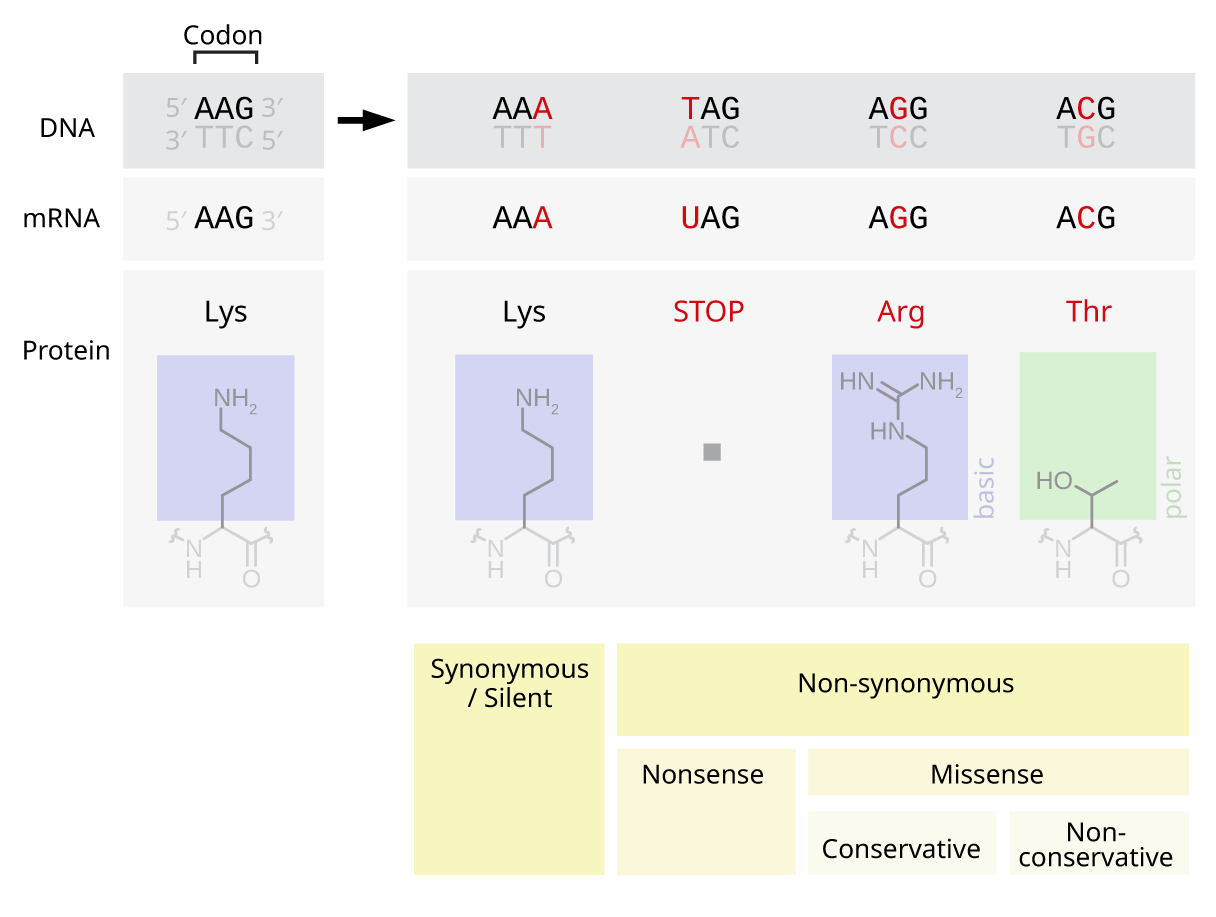
Genetic point mutation that results in an amino acid change in a protein
In genetics, a missense mutation is a point mutation in which a single nucleotide change results in a codon that codes for a different amino acid. It is a type of nonsynonymous substitution. Missense mutations change amino acids, which in turn alter proteins and may alter a protein's function or structure. These mutations may arise spontaneously from mutagens like UV radiation, tobacco smoke, an error in DNA replication, and other factors. Screening for missense mutations can be done by sequencing the genome of an organism and comparing the sequence to a reference genome to analyze for differences. Missense mutations can be repaired by the cell when there are errors in DNA replication by using mechanisms such as DNA proofreading and mismatch repair. They can also be repaired by using genetic engineering technologies or pharmaceuticals. Some notable examples of human diseases caused by missense mutations are Rett syndrome, cystic fibrosis, and sickle-cell disease.[[File:Missense Mutation Example.jpg|thumb|This image shows an example of missense mutation. One of the nucleotides (adenine) is replaced by another nucleotide (cytosine) in the DNA sequence. This results in an incorrect amino acid (proline) being incorporated into the protein sequence.|313x313px]]
Impact on protein function
Missense mutation refers to a change in one amino acid in a protein arising from a point mutation in a single nucleotide. Amino acids are the building blocks of proteins. Missense mutations are a type of nonsynonymous substitution in a DNA sequence. Two other types of nonsynonymous substitutions are nonsense mutations, in which a codon is changed to a premature stop codon that results in the resulting protein being cut short, and nonstop mutations, in which a stop codon deletion results in a longer but nonfunctional protein. The latter two types are not considered to be missense mutations.
Missense mutations can render the resulting protein nonfunctional, due to misfolding of the protein. These mutations are responsible for human diseases, such as Epidermolysis bullosa, sickle-cell disease, SOD1 mediated ALS, and a substantial number of cancers.
Not all missense mutations lead to appreciable protein changes. An amino acid may be replaced by a different amino acid of very similar chemical properties in which case the protein may still function normally; this is termed a conservative mutation. Alternatively, the amino acid substitution could occur in a region of the protein which does not significantly affect the protein secondary structure or function. Lastly, when more than one codon codes for the same amino acid (termed "degenerate coding"), the resulting mutation does not produce any change in translation and hence no change in protein is observed; degenerate coding would be classified as a synonymous substitution, or a silent mutation, and not a missense mutation.
Origin
Missense mutations may be inherited or arise spontaneously, termed de novo mutations. Well studied diseases arising from inherited missense mutations include sickle cell anemia, cystic fibrosis, and early-onset Alzheimer's and Parkinson's disease. De novo mutations that increase or decrease the activity of synapses have been implicated in the development of neurological and developmental disorders, such a Autism Spectrum Disorder and intellectual delay.
Agents of spontaneous missense mutation
Environmental mutagens, such as tobacco smoke or UV radiation, may be a cause of spontaneous missense mutations. Tobacco smoke has been implicated in transversion mutations in the K-ras gene, with a meta-analysis of lung carcinomas showing 25 tumours containing a G to T mutation causing an amino acid change from glycine to cysteine, and 11 tumours with a G to T mutation causing an amino acid change from glycine to valine. Similarly, numerous studies have shown ultraviolet light induces missense mutations in the p53 gene, which when unregulated, reduces the cell's ability to recognize DNA damage and engage in apoptosis, leading to cell proliferation and potential skin carcinogenesis.

DNA polymerase replication errors during cell division may lead to spontaneous missense mutations if DNA polymerase's proofreading ability does not detect and repair an error it makes. Spontaneous DNA polymerase errors are estimated to occur at a frequency of 1/109 base pairs.
Although rarer, tautomerization of bases also creates spontaneous missense mutations. Tautomerization occurs when hydrogen atoms on DNA bases spontaneously change locations, impacting the structure of the base, and allowing it to pair with an incorrect base. If this strand of DNA is replicated, the incorrect base will be the template for a new strand, leading to a mutation, possibly changing the amino acid and therefore, the protein. For example, Wang et al., (2011) used X-ray cystallography to demonstrate that a de novo mutation was created when DNA repair mechanisms did not recognize a C-A base mismatch due to tautomerization allowing the base structures to be compatible.
Screening
Next Generation Sequencing (NGS)
Next Generation Sequencing (NGS) has changed the world of sequencing by decreasing the cost of sequencing and increasing the throughput. It does this by utilizing massively parallel sequencing to sequence the genome. This involves clonally amplified DNA fragments that can be spatially separated into second generation sequencing (SGS) or third generation sequencing (TGS) platforms. There is variation between these protocols, but the overall methods are similar. Using massively parallel sequencing allows the NGS platform to produce very large sequences in a single run. The DNA fragments are typically separated by length using gel electrophoresis.
NGS consists of four main steps, DNA isolation, target enrichment, sequencing, and data analysis. The DNA isolation step involves breaking the genomic DNA into many small fragments. There are many different mechanisms that can be used to accomplish this such as mechanical methods, enzymatic digestion, and more. This step also consists of adding adaptors to either end of the DNA fragments that are complementary to the flow cell oligos and include primer binding sites for the target DNA. The target enrichment step amplifies the region of interest. This includes creating a complementary strand to the DNA fragments through hybridization to a flow cell oligo. It then gets denatured and bridge amplification occurs before the reverse strand is finally washed and sequencing can occur. The sequencing step involves massive parallel sequencing of all DNA fragments simultaneously using a NGS sequencer. This information is saved and analyzed in the last step, data analysis, using bioinformatics software. This compares the sequences to a reference genome to align the fragments and show mutations in the targeted area of the sequence.
Newborn Screening (NBS)
Newborn screening (NBS) for missense mutations is increasingly incorporating genomic technologies in addition to traditional biochemical methods to improve the detection of genetic disorders early in life. Traditional NBS primarily relies on biochemical assays, such as tandem mass spectrometry, to detect metabolic abnormalities indicative of conditions like phenylketonuria or congenital hypothyroidism. However, these methods may miss genetic causes or produce ambiguous results. To address these deficiencies, next-generation sequencing (NGS) is being added to NBS programs. For instance, targeted gene panels and whole-exome sequencing (WES) are used to identify disease causing missense mutations in genes associated with treatable conditions, such as severe combined immunodeficiency (SCID) and cystic fibrosis. Studies like the BabyDetect project have demonstrated the utility of genomic screening in identifying disorders missed by conventional methods, with actionable results for conditions affecting more than 400 genes. In addition, genomic approaches allow for the detection of rare or recessive conditions that may not manifest biochemically at birth, significantly expanding the scope of diseases screened. These advancements align with the established principles of NBS, which emphasize early detection and intervention to prevent morbidity and mortality.
Prevention and repair mechanisms

Cellular mechanisms
DNA polymerases, used in DNA replication, have a high specificity of 104 to 106-fold in base pairing. They have proofreading abilities to correct incorrect matches, allowing 90-99.9% of mismatches to be excised and repaired. The base mismatches that go unnoticed are repaired by the DNA mismatch repair pathway, also inherent in cells. The DNA mismatch repair pathway uses exonucleases that move along the DNA strand and remove the incorrectly incorporated base in order for DNA polymerase to fill in the correct base.Exonuclease1 is involved in many DNA repair systems and moves 5' to 3' on the DNA strand.
Genetic engineering and drug-based interventions
More recently, research has explored the use of genetic engineering and pharmaceuticals as potential treatments. tRNA therapies have emerged in research studies as a potential missense mutation treatment, following evidence supporting their use in nonsense mutation correction. Missense-correcting tRNAs are engineered to identify the mutated codon, but carry the correct charged amino acid which is inserted into the nascent protein.
Pharmaceuticals that target specific proteins affected by missense mutations have also shown therapeutic potential. Pharmaceutical studies have particularly focused on targeting the p53 mutant protein and Ca2+ channel abnormalities, both caused by gain of function missense mutations due to their high prevalence in a number of cancers and genetic diseases respectively. In cystic fibrosis, most commonly caused by missense mutations, drugs known as modulators target the defective Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) protein. For example, to reduce the defects caused by class III CFTR mutations, Ivacaftor, part of the modulator Kalydeco, forces the chloride channel to remain in an open position.
Future technology and research
Gene therapy is being explored as a treatment for missense mutations. This involves inserting the correct sequence of DNA into an incorrect gene. Artificial Intelligence programs, such as AlphaFold, are also being developed to predict the effect of missense mutations. Identifying potential deleterious mutations can assist with disease diagnosis and treatment.
Evolution

If a missense mutation is not deleterious, it will not be selected against and can contribute to species divergence. Over time, mutations occur randomly in individuals and can become fixed in populations if they are not selected against. Missense mutations are a type of mutation that are not neutral, and therefore can be acted on by selection. Selection cannot act on synonymous mutations (mutations that do not change anything phenotypically).
Tracking missense mutations, like nonsynonymous SNPs, in ancestral species populations allow genealogies and phylogenetic trees to be created and evolutionary connections to be made. Missense mutation analysis is often used in evolutionary genetics to create relationships between species, as amino acid changes leading to protein changes are needed for species to diverge from each other.
Notable examples
LMNA
_mutation_R527L_PMID_22549407.png)
DNA: 5' - AAC AGC CTG CGT ACG GCT CTC - 3' 3' - TTG TCG GAC GCA TGC CGA GAG - 5' mRNA: 5' - AAC AGC CUG CGU ACG GCU CUC - 3' Protein: Asn Ser Leu Arg Thr Ala Leu
LMNA missense mutation (c.1580GT) introduced at LMNA gene – position 1580 (nt) in the DNA sequence (CGT) causing the guanine to be replaced with the thymine, yielding CTT in the DNA sequence. This results at the protein level in the replacement of the arginine by the leucine at the position 527. This leads to destruction of salt bridge and structure destabilization. At phenotype level this manifests with overlapping mandibuloacral dysplasia and progeria syndrome.
The resulting transcript and protein product is:
DNA: 5' - AAC AGC CTG CTT ACG GCT CTC - 3' 3' - TTG TCG GAC GAA TGC CGA GAG - 5' mRNA: 5' - AAC AGC CUG CUU ACG GCU CUC - 3' Protein: Asn Ser Leu Leu Thr Ala Leu
Rett Syndrome
Missense mutations in the MeCP2 protein can cause Rett syndrome, otherwise known as the RTT phenotype. This phenotype primarily effects females, as males do not live with this mutation past infancy. T158M, R306C and R133C are the most common missense mutations causing RTT. T158M is a mutation of an adenine being substituted for a guanine causing the threonine at amino acid position 158 being substituted with a methionine. R133C is a mutation of a cytosine at base position 417 in the gene encoding the MeCP2 protein being substituted for a thymine, causing an amino acid substitution at position 133 in the protein of arginine with cysteine.
Sickle Cell
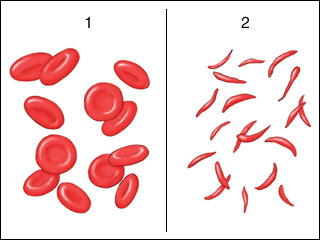
Other conditions that can be caused by missense mutations
- Alzheimers
- X-linked intellectual disability
- Hypocholesterolemia
- Tangier disease
- Congenital nemaline myopathy
References
References
- (2012-03-19). "Definition of Missense mutation". MedicineNet.
- Brown, TA. (2002). "Genomes. 2nd edition.". Oxford: Wiley-Liss.
- (2019-04-16). "Nonsynonymous, synonymous and nonsense mutations in human cancer-related genes undergo stronger purifying selections than expectation". BMC Cancer.
- (2025-02-14). "Nonstop mutations cause loss of renal tumor suppressor proteins VHL and BAP1 and affect multiple stages of protein translation". Science Advances.
- (August 2011). "Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer?". Molecular Cancer.
- (November 2013). "Molecular mechanisms of disease-causing missense mutations". Journal of Molecular Biology.
- (September 2021). "Management of Cutaneous Manifestations of Genetic Epidermolysis Bullosa: A Multiple Case Series". Journal of Wound, Ostomy, and Continence Nursing.
- (April 2017). "Sickle Cell Disease". The New England Journal of Medicine.
- (October 2006). "ALS: a disease of motor neurons and their nonneuronal neighbors". Neuron.
- (May 1, 2020). "A Monumental Breakthrough?". The News-Star.
- (2007-01-26). "A "Silent" Polymorphism in the MDR 1 Gene Changes Substrate Specificity". Science.
- (2017-06-01). "Ion channelopathies associated genetic variants as the culprit for sudden unexplained death". Forensic Science International.
- (2025). "Genetics, DNA Damage and Repair". StatPearls Publishing.
- (2024-08-29). "Identification of genetic variants associated with clinical features of sickle cell disease". Scientific Reports.
- (1993). "General Cystic Fibrosis Mutations Are Usually Missense Mutations Affecting Two Specific Protein Domains and Associated with a Specific RFLP Marker Haplotype". European Journal of Human Genetics.
- (March 2021). "Contribution of homozygous and compound heterozygous missense mutations in VWA2 to Alzheimer's disease". Neurobiology of Aging.
- (2019-04-09). "SNCA G51D Missense Mutation Causing Juvenile Onset Parkinson's Disease (P5.8-026)". Neurology.
- (2017-08-01). "Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains". Nature Neuroscience.
- (2001-09-15). "Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung". Cancer.
- (2024-11-27). "P53 and the Ultraviolet Radiation-Induced Skin Response: Finding the Light in the Darkness of Triggered Carcinogenesis". Cancers.
- (April 2009). "Ultraviolet A within Sunlight Induces Mutations in the Epidermal Basal Layer of Engineered Human Skin". The American Journal of Pathology.
- (2016-11-10). "Next generation sequencing of benzo(a)pyrene-induced lacZ mutants identifies a germ cell-specific mutation spectrum". Scientific Reports.
- Shen, Chang-Hui. (2023-01-01). "Chapter 1 - Nucleic acids: DNA and RNA". Academic Press.
- Shen, Chang-Hui. (2023-01-01). "Chapter 2 - Nucleic acid-based cellular activities – DNA replication, damage, and repair". Academic Press.
- (2011-10-25). "Structural evidence for the rare tautomer hypothesis of spontaneous mutagenesis". Proceedings of the National Academy of Sciences.
- (2013-12-01). "Clinical application of amplicon-based next-generation sequencing in cancer". Cancer Genetics.
- (2014). "Next-generation sequencing: current technologies and {{sic". Caister academic press.
- (2013). "Next Generation Sequencing Technologies in Medical Genetics". Springer New York.
- (February 2019). "Next-generation sequencing and its clinical application". Cancer Biology & Medicine.
- Levy, Harvey L. (1998-12-01). "Newborn Screening by Tandem Mass Spectrometry: A New Era". Clinical Chemistry.
- Cunningham, George. (2002-04-04). "The Science and Politics of Screening Newborns". New England Journal of Medicine.
- (2021-05-26). "Next-Generation Sequencing in Newborn Screening: A Review of Current State". Frontiers in Genetics.
- (January 2025). "Population-based, first-tier genomic newborn screening in the maternity ward". Nature Medicine.
- (2021). "Neonatal Hematology: Pathogenesis, Diagnosis, and Management of Hematologic Problems". Cambridge University Press.
- (September 2023). "Genome Sequencing for Newborn Screening-An Effective Approach for Tackling Rare Diseases". JAMA Network Open.
- (2005). "A Clinical Guide to Inherited Metabolic Diseases.". Cambridge University Press.
- Scharer, O. D.. (2013-10-01). "Nucleotide Excision Repair in Eukaryotes". Cold Spring Harbor Perspectives in Biology.
- (April 2004). "DNA replication fidelity". The Journal of Biological Chemistry.
- (January 2008). "Mechanisms and functions of DNA mismatch repair". Cell Research.
- (2005-06-01). "DNA mismatch repair". Annual Review of Biochemistry.
- (August 2015). "Exonuclease 1-dependent and independent mismatch repair". DNA Repair.
- (February 2024). "Engineered mischarged transfer RNAs for correcting pathogenic missense mutations". Molecular Therapy.
- (2021-03-03). "Voltage-Gated Ca2+-Channel α1-Subunit de novo Missense Mutations: Gain or Loss of Function - Implications for Potential Therapies". Frontiers in Synaptic Neuroscience.
- (June 2018). "Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target". Cancers.
- (June 2021). "Repurposing tRNAs for nonsense suppression". Nature Communications.
- (2017-08-11). "General Cystic Fibrosis Mutations Are Usually Missense Mutations Affecting Two Specific Protein Domains and Associated with a Specific RFLP Marker Haplotype". European Journal of Human Genetics.
- (2016-05-01). "Current and future treatment options for cystic fibrosis lung disease: latest evidence and clinical implications". Therapeutic Advances in Chronic Disease.
- "CFTR Modulator Therapies {{!}} Cystic Fibrosis Foundation".
- (2023-09-22). "Accurate proteome-wide missense variant effect prediction with AlphaMissense". Science.
- (April 2007). "Most Rare Missense Alleles Are Deleterious in Humans: Implications for Complex Disease and Association Studies". The American Journal of Human Genetics.
- (July 2003). "A phylogenetic approach to assessing the significance of missense mutations in disease genes". Human Mutation.
- (December 2010). "Triangulation of the human, chimpanzee, and Neanderthal genome sequences identifies potentially compensated mutations". Human Mutation.
- (2022-06-23). "Synonymous mutations in representative yeast genes are mostly strongly non-neutral". Nature.
- (2024-10-25). "Forward genetic approach identifies a phylogenetically conserved serine residue critical for the catalytic activity of UBIQUITIN-SPECIFIC PROTEASE 12 in Arabidopsis". Scientific Reports.
- (May 2015). "Long-Term Balancing Selection in LAD1 Maintains a Missense Trans-Species Polymorphism in Humans, Chimpanzees, and Bonobos". Molecular Biology and Evolution.
- (November 2012). "A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome". European Journal of Human Genetics.
- (February 2016). "The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome". Human Molecular Genetics.
- (2014). "Comprehensive Guide to Autism". Springer New York.
- (October 1999). "Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2". Nature Genetics.
- (2021-03-01). "Evolutionary history of sickle-cell mutation: implications for global genetic medicine". Human Molecular Genetics.
- "141900 Hemoglobin—Beta Locus; HBB: .0243 Hemoglobin S. Sickle Cell Anemia, included. Malaria, Resistance to, included. HBB, GLU6VAL — 141900.0243". [[Mendelian Inheritance in Man.
- (1990-04-27). "Sickle Cell Disease. Charles F. Whrrren and John F. Bertles, Eds. New York Academy of Sciences, New York, 1989. xiv, 477 pp., illus. $119. Annals of the New York Academy of Sciences, vol. 565. From a conference, Bethesda, MD, April 1988". Science.
- (August 2016). "The de novo missense mutation N117S in skeletal muscle α-actin 1 causes a mild form of congenital nemaline myopathy". Molecular Medicine Reports.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about Missense mutation — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report