From Surf Wiki (app.surf) — the open knowledge base
Metal carbonyl
Coordination complexes of transition metals with carbon monoxide ligands

Coordination complexes of transition metals with carbon monoxide ligands

Metal carbonyls are coordination complexes of transition metals with carbon monoxide ligands. Metal carbonyls are useful in organic synthesis and as catalysts or catalyst precursors in homogeneous catalysis, such as hydroformylation and Reppe chemistry. In the Mond process, nickel tetracarbonyl is used to produce pure nickel. In organometallic chemistry, metal carbonyls serve as precursors for the preparation of other organometallic complexes.
Metal carbonyls are toxic by skin contact, inhalation or ingestion, in part because of their ability to carbonylate hemoglobin to give carboxyhemoglobin, which prevents the binding of oxygen.
Nomenclature and terminology
The nomenclature of the metal carbonyls depends on the charge of the complex, the number and type of central atoms, and the number and type of ligands and their binding modes. They occur as neutral complexes, as positively-charged metal carbonyl cations or as negatively charged metal carbonylates. The carbon monoxide ligand may be bound terminally to a single metal atom or bridging to two or more metal atoms. These complexes may be homoleptic, containing only CO ligands, such as nickel tetracarbonyl (Ni(CO)4), but more commonly metal carbonyls are heteroleptic and contain a mixture of ligands.
Mononuclear metal carbonyls contain only one metal atom as the central atom. Except vanadium hexacarbonyl, only metals with even atomic number, such as chromium, iron, nickel, and their homologs, build neutral mononuclear complexes. Polynuclear metal carbonyls are formed from metals with odd atomic numbers and contain a metal–metal bond. Complexes with different metals but only one type of ligand are called isoleptic.
Carbon monoxide has distinct binding modes in metal carbonyls. They differ in terms of their hapticity, denoted η, and their bridging mode. In η2-CO complexes, both the carbon and oxygen are bonded to the metal. More commonly only carbon is bonded, in which case the hapticity is not mentioned.
The carbonyl ligand engages in a wide range of bonding modes in metal carbonyl dimers and clusters. In the most common bridging mode, denoted μ2 or simply μ, the CO ligand bridges a pair of metals. This bonding mode is observed in the commonly available metal carbonyls: Co2(CO)8, Fe2(CO)9, Fe3(CO)12, and Co4(CO)12.{{cite book
:[[File:CObondingmodes2.jpg|class=skin-invert-image]]
Structure and bonding


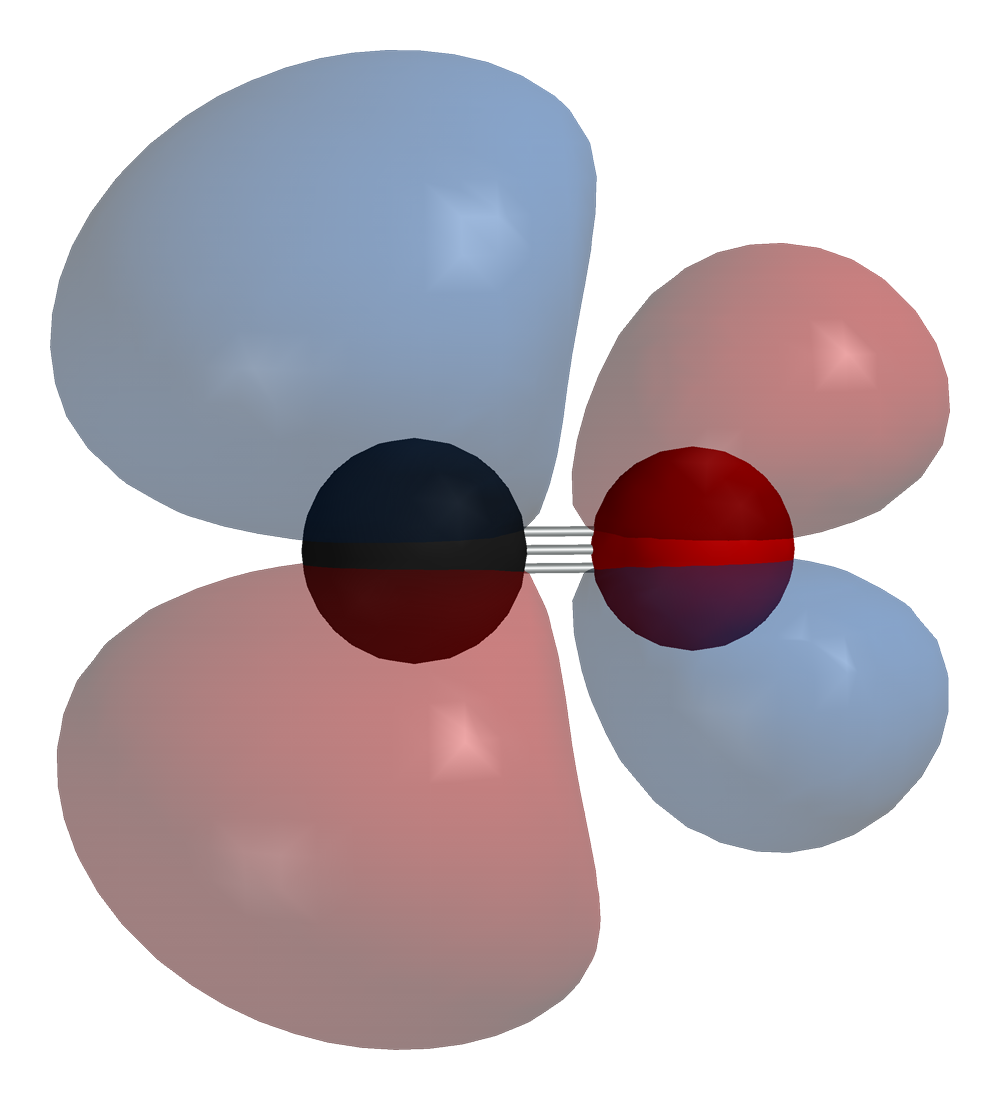

Carbon monoxide bonds to transition metals using "synergistic pi* back-bonding". The M–C bonding has three components, giving rise to a partial triple bond. A sigma (σ) bond arises from overlap of the nonbonding (or weakly anti-bonding) sp-hybridized electron pair on carbon with a blend of d-, s-, and p-orbitals on the metal. A pair of pi (π) bonds arises from overlap of filled d-orbitals on the metal with a pair of π*-antibonding orbitals projecting from the carbon atom of the CO. The latter kind of binding requires that the metal have d-electrons, and that the metal be in a relatively low oxidation state (0 or +1) which makes the back-donation of electron density favorable. As electrons from the metal fill the π-antibonding orbital of CO, they weaken the carbon–oxygen bond compared with free carbon monoxide, while the metal–carbon bond is strengthened. Because of the multiple bond character of the M–CO linkage, the distance between the metal and carbon atom is relatively short, often less than 1.8 Å, about 0.2 Å shorter than a metal–alkyl bond. The M-CO and MC-O distance are sensitive to other ligands on the metal. Illustrative of these effects are the following data for Mo-C and C-O distances in Mo(CO)6 and Mo(CO)3(4-methylpyridine)3: 2.06 vs 1.90 and 1.11 vs 1.18 Å.
Infrared spectroscopy is a sensitive probe for the presence of bridging carbonyl ligands. For compounds with doubly bridging CO ligands, denoted μ2-CO or often just μ-CO, the bond stretching frequency νCO is usually shifted by 100–200 cm−1 to lower energy compared to the signatures of terminal CO, which are in the region 1800 cm−1. Bands for face-capping (μ3) CO ligands appear at even lower energies. In addition to symmetrical bridging modes, CO can be found to bridge asymmetrically or through donation from a metal d orbital to the π* orbital of CO.{{cite book
Physical characteristics
Most mononuclear carbonyl complexes are colorless or pale yellow, volatile liquids or solids that are flammable and toxic. Vanadium hexacarbonyl, a uniquely stable 17-electron metal carbonyl, is a blue-black solid.
Analytical characterization

Apart from X-ray crystallography, important analytical techniques for the characterization of metal carbonyls are infrared spectroscopy and 13C NMR spectroscopy. These two techniques provide structural information on two very different time scales. Infrared-active vibrational modes, such as CO-stretching vibrations, are often fast compared to intramolecular processes, whereas NMR transitions occur at lower frequencies and thus sample structures on a time scale that, it turns out, is comparable to the rate of intramolecular ligand exchange processes. NMR data provide information on "time-averaged structures", whereas IR is an instant "snapshot".{{cite journal

Iron pentacarbonyl exhibits only a single 13C NMR signal owing to rapid exchange of the axial and equatorial CO ligands by Berry pseudorotation.
Infrared spectra

An important technique for characterizing metal carbonyls is infrared spectroscopy.{{cite book
The number of vibrational modes of a metal carbonyl complex can be determined by group theory. Only vibrational modes that transform as the electric dipole operator will have nonzero direct products and are observed. The number of observable IR transitions (but not their energies) can thus be predicted.{{cite book
| Compound | νCO (cm−1) | 13C NMR shift (ppm) | average M-CO distance (pm) | average C-O distance (pm) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CO | 2143 | 181 | ||||||||||
| 1748 | 245 | 204 | 116 | |||||||||
| 1859 | (paramagnetic) | 200, 193 (PPN+ salt) | 113 | |||||||||
| Cr(CO)6 | 2000 | 212 | 191 | 114 | ||||||||
| 2100 | ||||||||||||
| 2204 | 191 | 112 (BF4− salt) | ||||||||||
| Fe(CO)5 | 2022, 2000 | 209 | 180 | 112 | ||||||||
| Ru(CO)5 | author = Adams R. D., Barnard T. S., Cortopassi J. E., Wu W., Li Z. | title = Inorganic Syntheses | year = 1998 | chapter = Platinum-ruthenium carbonyl cluster complexes | series = Inorganic Syntheses | volume = 32 | pages = 280–284 | doi = 10.1002/9780470132630.ch44 | isbn = 978-0-470-13263-0 }} | |||
| Ni(CO)4 | 181 | 113 |
| Carbonyl | νCO, μ1 (cm−1) | νCO, μ2 (cm−1) | νCO, μ3 (cm−1) |
|---|---|---|---|
| Rh2(CO)8 | 2060, 2084 | 1846, 1862 | |
| Rh4(CO)12 | 2044, 2070, 2074 | 1886 | |
| Rh6(CO)16 | 2045, 2075 | 1819 |
Nuclear magnetic resonance spectroscopy
Metal carbonyls are often characterized by 13C NMR spectroscopy. To improve the sensitivity of this technique, complexes are often enriched with 13CO. Typical chemical shift range for terminally bound ligands is 150 to 220 ppm. Bridging ligands resonate between 230 and 280 ppm.{{cite book
NMR spectroscopy can be used for experimental determination of the fluxionality.
The activation energy of ligand exchange processes can be determined by the temperature dependence of the line broadening.{{cite book
Mass spectrometry
Mass spectrometry provides information about the structure and composition of the complexes. Spectra for metal polycarbonyls are often easily interpretable, because the dominant fragmentation process is the loss of carbonyl ligands (m/z = 28).
: → + CO
Electron ionization is the most common technique for characterizing the neutral metal carbonyls. Neutral metal carbonyls can be converted to charged species by derivatization, which enables the use of electrospray ionization (ESI), instrumentation for which is often widely available. For example, treatment of a metal carbonyl with alkoxide generates an anionic metallaformate that is amenable to analysis by ESI-MS:
:LnM(CO) + RO− → [LnM−C(=O)OR]−
Some metal carbonyls react with azide to give isocyanato complexes with release of nitrogen.{{cite book
Mass spectrometry combined with infrared photodissociation spectroscopy can provide vibrational informations for ionic carbonyl complexes in gas phase.
Occurrence in nature

|doi-access= free In the investigation of the infrared spectrum of the Galactic Center of the Milky Way, monoxide vibrations of iron carbonyls in interstellar dust clouds were detected.{{cite journal |doi-access= free
In the oxygen-rich atmosphere of the Earth, metal carbonyls are subject to oxidation to the metal oxides. It is discussed whether in the reducing hydrothermal environments of the prebiotic prehistory such complexes were formed and could have been available as catalysts for the synthesis of critical biochemical compounds such as pyruvic acid.{{cite journal
The hydrogenase enzymes contain CO bound to iron. It is thought that the CO stabilizes low oxidation states, which facilitates the binding of hydrogen. The enzymes carbon monoxide dehydrogenase and acetyl-CoA synthase also are involved in bioprocessing of CO.{{cite book
Synthesis
The synthesis of metal carbonyls is a widely studied subject of organometallic research. Since the work of Mond and then Hieber, many procedures have been developed for the preparation of mononuclear metal carbonyls as well as homo- and heterometallic carbonyl clusters.
Direct reaction of metal with carbon monoxide
Nickel tetracarbonyl and iron pentacarbonyl can be prepared according to the following equations by reaction of finely divided metal with carbon monoxide:{{cite book
:Ni + 4 CO → Ni(CO)4 (1 bar, 55 °C) :Fe + 5 CO → Fe(CO)5 (100 bar, 175 °C)
Nickel tetracarbonyl is formed with carbon monoxide already at 80 °C and atmospheric pressure, finely divided iron reacts at temperatures between 150 and 200 °C and a carbon monoxide pressure of 50–200 bar. Other metal carbonyls are prepared by less direct methods.
Reduction of metal salts and oxides
Some metal carbonyls are prepared by the reduction of metal halides in the presence of high pressure of carbon monoxide. A variety of reducing agents are employed, including copper, aluminum, hydrogen, as well as metal alkyls such as triethylaluminium. Illustrative is the formation of chromium hexacarbonyl from anhydrous chromium(III) chloride in benzene with aluminum as a reducing agent, and aluminum chloride as the catalyst:
:CrCl3 + Al + 6 CO → Cr(CO)6 + AlCl3
The use of metal alkyls, such as triethylaluminium and diethylzinc, as the reducing agent leads to the oxidative coupling of the alkyl radical to form the dimer alkane:
:WCl6 + 6 CO + 2 Al(C2H5)3 → W(CO)6 + 2 AlCl3 + 3 C4H10
Tungsten, molybdenum, manganese, and rhodium salts may be reduced with lithium aluminium hydride. Vanadium hexacarbonyl is prepared with sodium as a reducing agent in chelating solvents such as diglyme.{{cite book
:VCl3 + 4 Na + 6 CO + 2 diglyme → Na(diglyme)2[V(CO)6] + 3 NaCl :[V(CO)6]− + H+ → H[V(CO)6] → H2 + V(CO)6
In the aqueous phase, nickel or cobalt salts can be reduced, for example by sodium dithionite. In the presence of carbon monoxide, cobalt salts are quantitatively converted to the tetracarbonylcobalt(−1) anion:
:Co2+ + + 6 OH− + 4 CO → + 3 + 3 H2O
Some metal carbonyls are prepared using CO directly as the reducing agent. In this way, Hieber and Fuchs first prepared dirhenium decacarbonyl from the oxide:{{cite journal
:Re2O7 + 17 CO → Re2(CO)10 + 7 CO2
If metal oxides are used carbon dioxide is formed as a reaction product. In the reduction of metal chlorides with carbon monoxide phosgene is formed, as in the preparation of osmium carbonyl chloride from the chloride salts. Carbon monoxide is also suitable for the reduction of sulfides, where carbonyl sulfide is the byproduct.
Photolysis and thermolysis
Photolysis or thermolysis of mononuclear carbonyls generates di- and polymetallic carbonyls such as diiron nonacarbonyl (Fe2(CO)9).{{cite book
:2 Fe(CO)5 → Fe2(CO)9 + CO
The thermal decomposition of triosmium dodecacarbonyl (Os3(CO)12) provides higher-nuclear osmium carbonyl clusters such as Os4(CO)13, Os6(CO)18 up to Os8(CO)23.
Mixed ligand carbonyls of ruthenium, osmium, rhodium, and iridium are often generated by abstraction of CO from solvents such as dimethylformamide (DMF) and 2-methoxyethanol. Typical is the synthesis of IrCl(CO)(PPh3)2 from the reaction of iridium(III) chloride and triphenylphosphine in boiling DMF solution.
Salt metathesis
Salt metathesis reaction of salts such as KCo(CO)4 with [Ru(CO)3Cl2]2 leads selectively to mixed-metal carbonyls such as RuCo2(CO)11.{{cite journal
:4 KCo(CO)4 + [Ru(CO)3Cl2]2 → 2 RuCo2(CO)11 + 4 KCl + 11 CO
Cations and carbonylates
The synthesis of ionic carbonyl complexes is possible by oxidation or reduction of the neutral complexes. Anionic metal carbonylates can be obtained for example by reduction of dinuclear complexes with sodium. A familiar example is the sodium salt of iron tetracarbonylate (Na2Fe(CO)4, Collman's reagent), which is used in organic synthesis.{{cite book
The cationic hexacarbonyl salts of manganese, technetium and rhenium can be prepared from the carbonyl halides under carbon monoxide pressure by reaction with a Lewis acid. :Mn(CO)5Cl + AlCl3 + CO → [][]
The use of strong acids succeeded in preparing gold carbonyl cations such as [Au(CO)2]+, which is used as a catalyst for the carbonylation of alkenes.{{cite journal |hdl-access= free
Reactions
Metal carbonyls are important precursors for the synthesis of other organometallic complexes. Common reactions are the substitution of carbon monoxide by other ligands, the oxidation or reduction reactions of the metal center, and reactions at the carbon monoxide ligand.
CO substitution
The substitution of CO ligands can be induced thermally or photochemically by donor ligands. The range of ligands is large, and includes phosphines, cyanide (CN−), nitrogen donors, and even ethers, especially chelating ones. Alkenes, especially dienes, are effective ligands that afford synthetically useful derivatives. Substitution of 18-electron complexes generally follows a dissociative mechanism, involving 16-electron intermediates.
Substitution proceeds via a dissociative mechanism: :M(CO)n → M(CO)n−1 + CO :M(CO)n−1 + L → M(CO)n−1L
The dissociation energy is 105 kJ/mol for nickel tetracarbonyl and 155 kJ/mol for chromium hexacarbonyl.
Substitution in 17-electron complexes, which are rare, proceeds via associative mechanisms with a 19-electron intermediates. :M(CO)n + L → M(CO)nL :M(CO)nL → M(CO)n−1L + CO
The rate of substitution in 18-electron complexes is sometimes catalysed by catalytic amounts of oxidants, via electron transfer.{{cite journal
Reduction
Metal carbonyls react with reducing agents such as metallic sodium or sodium amalgam to give carbonylmetalate (or carbonylate) anions. Polynuclear or electron-imprecise clusters add the reductant to give mononuclear anions: :Mn2(CO)10 + 2 Na → 2 Na[Mn(CO)5] Conversely, electron-precise mononuclear compounds lose CO and may form clusters: :Fe(CO)5 + 2 Na → Na2[Fe(CO)4] + CO :2 Cr(CO)6 + 2 Na → Na2Cr2(CO)10
Mercury can insert into the metal–metal bonds of some polynuclear metal carbonyls: :Co2(CO)8 + Hg → (CO)4Co−Hg−Co(CO)4
Nucleophilic attack at CO
The CO ligand is often susceptible to attack by nucleophiles. For example, trimethylamine oxide and potassium bis(trimethylsilyl)amide convert CO ligands to CO2 and CN−, respectively. In the "Hieber base reaction", hydroxide ion attacks the CO ligand to give a metallacarboxylic acid, followed by the release of carbon dioxide and the formation of metal hydrides or carbonylmetalates. A well-studied example of this nucleophilic addition is the conversion of iron pentacarbonyl to hydridoiron tetracarbonyl anion: :Fe(CO)5 + NaOH → Na[Fe(CO)4CO2H] :Na[Fe(CO)4COOH] + NaOH → Na[HFe(CO)4] + NaHCO3
Hydride reagents also attack CO ligands, especially in cationic metal complexes, to give the formyl derivative: :[Re(CO)6]+ + H− → Re(CO)5CHO
Organolithium reagents add with metal carbonyls to acylmetal carbonyl anions. O-Alkylation of these anions, such as with Meerwein salts, affords Fischer carbenes.

With electrophiles
Despite being in low formal oxidation states, metal carbonyls are relatively unreactive toward many electrophiles. For example, they resist attack by alkylating agents, mild acids, and mild oxidizing agents. Most metal carbonyls do undergo halogenation. Iron pentacarbonyl, for example, forms ferrous carbonyl halides: :Fe(CO)5 + X2 → Fe(CO)4X2 + CO
Metal–metal bonds are cleaved by halogens. Depending on the electron-counting scheme used, this can be regarded as an oxidation of the metal atoms: :Mn2(CO)10 + Cl2 → 2 Mn(CO)5Cl
Compounds
Most metal carbonyl complexes contain a mixture of ligands. Examples include the historically important IrCl(CO)(P(C6H5)3)2 and the antiknock agent (CH3C5H4)Mn(CO)3. The parent compounds for many of these mixed ligand complexes are the binary carbonyls, those species of the formula [Mx(CO)n]z, many of which are commercially available. The formulae of many metal carbonyls can be inferred from the 18-electron rule.
Binary, charge-neutral
- Group 2 elements calcium, strontium, and barium can all form octacarbonyl complexes M(CO)8 (M = Ca, Sr, Ba). The compounds were characterized in cryogenic matrices by vibrational spectroscopy and in gas phase by mass spectrometry.
- Group 4 elements with 4 valence electrons are expected to form heptacarbonyls; while these are extremely rare, substituted derivatives of Ti(CO)7 are known.
- Group 5 elements with 5 valence electrons, again are subject to steric effects that prevent the formation of M–M bonded species such as V2(CO)12, which is unknown. The 17-VE V(CO)6 is however well known.
- Group 6 elements with 6 valence electrons form hexacarbonyls Cr(CO)6, Mo(CO)6, W(CO)6, and Sg(CO)6. Group 6 elements (as well as group 7) are also well known for exhibiting the cis effect (the labilization of CO in the cis position) in organometallic synthesis.
- Group 7 elements with 7 valence electrons form pentacarbonyl dimers Mn2(CO)10, Tc2(CO)10, and Re2(CO)10.
- Group 8 elements with 8 valence electrons form pentacarbonyls Fe(CO)5, Ru(CO)5 and Os(CO)5. The heavier two members are unstable, tending to decarbonylate to give Ru3(CO)12, and Os3(CO)12. The two other principal iron carbonyls are Fe3(CO)12 and Fe2(CO)9.
- Group 9 elements with 9 valence electrons and are expected to form tetracarbonyl dimers M2(CO)8. In fact the cobalt derivative of this octacarbonyl is the only stable member, but all three tetramers are well known: Co4(CO)12, Rh4(CO)12, Rh6(CO)16, and Ir4(CO)12. Co2(CO)8 unlike the majority of the other 18 VE transition metal carbonyls is sensitive to oxygen.
- Group 10 elements with 10 valence electrons form tetracarbonyls such as Ni(CO)4. Curiously Pd(CO)4 and Pt(CO)4 are not stable.
Binary, anionic
- Group 3 elements scandium and yttrium, as well as lanthanum, form the 20-electron monoanions [Sc(CO)8]−, [Y(CO)8]−, and [La(CO)8]−.
- Group 4 elements as dianions resemble neutral group 6 derivatives: [Ti(CO)6]2−.{{cite journal
- Group 5 elements as monoanions resemble again neutral group 6 derivatives: [V(CO)6]−.
- Group 6 elements form (inter alia) anions with the lowest known oxidation state for transition metals: Cr(CO), Mo(CO), and W(CO).
- Group 7 elements as monoanions resemble neutral group 8 derivatives: [Mn(CO)5]−, [Tc(CO)5]−, [Re(CO)5]−.
- Group 8 elements as dianaions resemble neutral group 10 derivatives: [Fe(CO)4]2−, [Ru(CO)4]2−, [Os(CO)4]2−. Condensed derivatives are also known.
- Group 9 elements as monoanions resemble neutral group 10 metal carbonyl. [Co(CO)4]− is the best studied member.
Large anionic clusters of nickel, palladium, and platinum are also well known. Many metal carbonyl anions can be protonated to give metal carbonyl hydrides.
Binary, cationic
- Group 2 elements form [M(CO)8]+ (M = Ca, Sr, Ba), characterized in gas phase by mass spectrometry and vibrational spectroscopy.
- Group 3 elements form [Sc(CO)7]+ and [Y(CO)8]+ in gas phase.
- Group 7 elements as monocations resemble neutral group 6 derivative [M(CO)6]+ (M = Mn, Tc, Re).
- Group 8 elements as dications also resemble neutral group 6 derivatives [M(CO)6]2+ (M = Fe, Ru, Os).{{cite journal
Nonclassical
Nonclassical describes those carbonyl complexes where νCO is higher than that for free carbon monoxide. In nonclassical CO complexes, the C-O distance is shorter than free CO (113.7 pm). The structure of [Fe(CO)6]2+, with dC-O = 112.9 pm, illustrates this effect. These complexes are usually cationic, sometimes dicationic.
Applications

Metallurgical uses
Metal carbonyls are used in several industrial processes. Perhaps the earliest application was the extraction and purification of nickel via nickel tetracarbonyl by the Mond process (see also carbonyl metallurgy).
By a similar process carbonyl iron, a highly pure metal powder, is prepared by thermal decomposition of iron pentacarbonyl. Carbonyl iron is used inter alia for the preparation of inductors, pigments, as dietary supplements,{{cite journal
Catalysis
Metal carbonyls are used in a number of industrially important carbonylation reactions. In the oxo process, an alkene, hydrogen gas, and carbon monoxide react together with a catalyst (such as dicobalt octacarbonyl) to give aldehydes. Illustrative is the production of butyraldehyde from propylene: :CH3CH=CH2 + H2 + CO → CH3CH2CH2CHO
Butyraldehyde is converted on an industrial scale to 2-ethylhexanol, a precursor to PVC plasticizers, by aldol condensation, followed by hydrogenation of the resulting hydroxyaldehyde. The "oxo aldehydes" resulting from hydroformylation are used for large-scale synthesis of fatty alcohols, which are precursors to detergents. The hydroformylation is a reaction with high atom economy, especially if the reaction proceeds with high regioselectivity.
:[[File:Hydroformylation Mechanism V.1.svg|class=skin-invert-image|380px|Hydroformulation mechanism]]
Another important reaction catalyzed by metal carbonyls is the hydrocarboxylation. The example below is for the synthesis of acrylic acid and acrylic acid esters:
:[[File:Reppe-chemistry-carbonmonoxide-01.png|class=skin-invert-image|450px|Hydrocarboxylation of acetylene]]
:[[File:Reppe-chemistry-carbonmonoxide-02.png|class=skin-invert-image|450px|Hydrocarboxylation of acetylene with an alcohol]]
Also the cyclization of acetylene to cyclooctatetraene uses metal carbonyl catalysts:{{cite journal
In the Monsanto and Cativa processes, acetic acid is produced from methanol, carbon monoxide, and water using hydrogen iodide as well as rhodium and iridium carbonyl catalysts, respectively. Related carbonylation reactions afford acetic anhydride.
CO-releasing molecules (CO-RMs)
Carbon monoxide-releasing molecules are metal carbonyl complexes that are being developed as potential drugs to release CO. At low concentrations, CO functions as a vasodilatory and an anti-inflammatory agent. CO-RMs have been conceived as a pharmacological strategic approach to carry and deliver controlled amounts of CO to tissues and organs.
Toxicology
The toxicity of metal carbonyls is due to toxicity of carbon monoxide, the metal, and because of the volatility and instability of the complexes, any inherent toxicity of the metal is generally made much more severe due to ease of exposure. Exposure occurs by inhalation, or for liquid metal carbonyls by ingestion or due to the good fat solubility by skin resorption. Most clinical experience were gained from toxicological poisoning with nickel tetracarbonyl and iron pentacarbonyl due to their use in industry. Nickel tetracarbonyl is considered as one of the strongest inhalation poisons.{{cite book
Inhalation of nickel tetracarbonyl causes acute non-specific symptoms similar to a carbon monoxide poisoning, such as nausea, cough, headache, fever, and dizziness. After some time, severe pulmonary symptoms such as cough, tachycardia, and cyanosis, or problems in the gastrointestinal tract occur. In addition to pathological alterations of the lung, such as by metalation of the alveoli, damages are observed in the brain, liver, kidneys, adrenal glands, and spleen. A metal carbonyl poisoning often necessitates a lengthy recovery.{{cite book
Chronic exposure by inhalation of low concentrations of nickel tetracarbonyl can cause neurological symptoms such as insomnia, headaches, dizziness and memory loss. Nickel tetracarbonyl is considered carcinogenic, but it can take 20 to 30 years from the start of exposure to the clinical manifestation of cancer.{{cite book
History

Initial experiments on the reaction of carbon monoxide with metals were carried out by Justus von Liebig in 1834. By passing carbon monoxide over molten potassium he prepared a substance having the empirical formula KCO, which he called Kohlenoxidkalium.{{cite journal

The synthesis of the first true heteroleptic metal carbonyl complex was performed by Paul Schützenberger in 1868 by passing chlorine and carbon monoxide over platinum black, where dicarbonyldichloroplatinum (Pt(CO)2Cl2) was formed.{{cite journal
Ludwig Mond, one of the founders of Imperial Chemical Industries, investigated in the 1890s with Carl Langer and Friedrich Quincke various processes for the recovery of chlorine which was lost in the Solvay process by nickel metals, oxides, and salts. As part of their experiments the group treated nickel with carbon monoxide. They found that the resulting gas colored the gas flame of a burner in a greenish-yellowish color; when heated in a glass tube it formed a nickel mirror. The gas could be condensed to a colorless, water-clear liquid with a boiling point of 43 °C. Thus, Mond and his coworker had discovered the first pure, homoleptic metal carbonyl, nickel tetracarbonyl (Ni(CO)4).{{cite journal |author-link= Walter Gratzer |chapter-url= https://books.google.com/books?id=yDBep9y0SVsC
The following year, Mond and Marcellin Berthelot independently discovered iron pentacarbonyl, which is produced by a similar procedure as nickel tetracarbonyl. Mond recognized the economic potential of this class of compounds, which he commercially used in the Mond process and financed more research on related compounds. Heinrich Hirtz and his colleague M. Dalton Cowap synthesized metal carbonyls of cobalt, molybdenum, ruthenium, and diiron nonacarbonyl.{{cite journal In 1906 James Dewar and H. O. Jones were able to determine the structure of diiron nonacarbonyl, which is produced from iron pentacarbonyl by the action of sunlight.{{cite journal |doi-access= free}} After Mond, who died in 1909, the chemistry of metal carbonyls fell for several years in oblivion. BASF started in 1924 the industrial production of iron pentacarbonyl by a process which was developed by Alwin Mittasch. The iron pentacarbonyl was used for the production of high-purity iron, so-called carbonyl iron, and iron oxide pigment.{{cite journal
Walter Hieber played in the years following 1928 a decisive role in the development of metal carbonyl chemistry. He systematically investigated and discovered, among other things, the Hieber base reaction, the first known route to metal carbonyl hydrides and synthetic pathways leading to metal carbonyls such as dirhenium decacarbonyl.{{cite book

Also in the 1930s Walter Reppe, an industrial chemist and later board member of BASF, discovered a number of homogeneous catalytic processes, such as the hydrocarboxylation, in which olefins or alkynes react with carbon monoxide and water to form products such as unsaturated acids and their derivatives. In these reactions, for example, nickel tetracarbonyl or cobalt carbonyls act as catalysts.{{cite book

For the rational design of new complexes the concept of the isolobal analogy has been found useful. Roald Hoffmann was awarded the Nobel Prize in chemistry for the development of the concept. This describes metal carbonyl fragments of M(CO)n as parts of octahedral building blocks in analogy to the tetrahedral CH3–, CH2– or CH– fragments in organic chemistry. In example dimanganese decacarbonyl is formed in terms of the isolobal analogy of two d7 Mn(CO)5 fragments, that are isolobal to the methyl radical . In analogy to how methyl radicals combine to form ethane, these can combine to dimanganese decacarbonyl. The presence of isolobal analog fragments does not mean that the desired structures can be synthesized. In his Nobel Prize lecture Hoffmann emphasized that the isolobal analogy is a useful but simple model, and in some cases does not lead to success.{{cite web
The economic benefits of metal-catalysed carbonylations, such as Reppe chemistry and hydroformylation, led to growth of the area. Metal carbonyl compounds were discovered in the active sites of three naturally occurring enzymes.{{cite journal
References
References
- (2007). "Lehrbuch der Anorganischen Chemie". de Gruyter.
- Cotton, F. Albert. (1968). "Proposed nomenclature for olefin-metal and other organometallic complexes". Journal of the American Chemical Society.
- (1996). "The Crystal Structure of Tris(4-methylpyridine) Tricarbonylmolybdenum(0)". J Chem Crystallogr.
- (1966). "Inorganic Syntheses".
- (1990). "Highly reduced organometallics. 28. Synthesis, isolation, and characterization of [K(cryptand 2.2.2)]2[Hf(CO)6], the first substance to contain hafnium in a negative oxidation state. Structural characterization of [K(cryptand 2.2.2)]2[M(CO)6].cntdot.pyridine (M = Ti, Zr, and Hf)". American Chemical Society (ACS).
- (1979). "Crystal and Molecular Structure of Vanadium Hexacarbonyl". Acta Crystallographica.
- (1975-11-01). "Electronic structure of chromium hexacarbonyl at 78 K. I. Neutron diffraction study". International Union of Crystallography (IUCr).
- (2005-05-10). "Homoleptic, σ-Bonded Octahedral Superelectrophilic Metal Carbonyl Cations of Iron(II), Ruthenium(II), and Osmium(II). Part 2: Syntheses and Characterizations of [M(CO)6][BF4]2 (M = Fe, Ru, Os)". American Chemical Society (ACS).
- (1993). "Nickel carbonyl [Ni(CO)4] and iron carbonyl [Fe(CO)5]: molecular structures in the solid state". American Chemical Society (ACS).
- Adams R. D., Barnard T. S., Cortopassi J. E., Wu W., Li Z.. (1998). "Inorganic Syntheses".
- Elliot. Band. (1978). "Mechanistic features of metal cluster rearrangements". Chem. Rev..
- (2011). "Infrared spectroscopy of mass-selected metal carbonyl cations". Journal of Molecular Spectroscopy.
- Miessler, Gary L.. (2013). "Inorganic Chemistry". Prentice Hall.
- (1999). "Synthesis and Technique in Inorganic Chemistry". University Science Books.
- (2018). "A Ferrocene-Based Dicationic Iron(IV) Carbonyl Complex". Angewandte Chemie International Edition.
- Jim D. Atwood. (1997). "Inorganic and Organometallic Reaction Mechanisms". Wiley.
- Ellis, J. E.. (2003). "Metal carbonyl anions". [[Organometallics]].
- (2018-08-31). "Observation of alkaline earth complexes M(CO)8 (M = Ca, Sr, or Ba) that mimic transition metals". Science.
- (2018-04-25). "Octacarbonyl Anion Complexes of Group Three Transition Metals [TM(CO)8]− (TM = Sc, Y, La) and the 18-Electron Rule". Angewandte Chemie International Edition.
- Ellis, John E.. (2006-04-17). "Adventures with substances containing metals in negative oxidation states". Inorganic Chemistry.
- (2013). "Testing the Limits of the 18-Electron Rule: The Gas-Phase Carbonyls of Sc+ and Y+". Inorganic Chemistry.
- (2020). "The Nature of Nonclassical Carbonyl Ligands Explained by Kohn–Sham Molecular Orbital Theory". Chemistry – A European Journal.
- Hartwig, John. (2010). "Organotransition Metal Chemistry: From Bonding to Catalysis". University Science Books.
- Motterlini Roberto, Otterbein Leo. (2010). "The therapeutic potential of carbon monoxide". Nature Reviews Drug Discovery.
- (1980). "Carbonyl, Thiocarbonyl, Selenocarbonyl, and Tellurocarbonyl Complexes Derived from a Dichlorocarbene Complex of Osmium". Journal of the American Chemical Society.
- D. Lentz. (1994). "Fluorinated Isocyanides - More than Ligands with Unusual Properties". Angewandte Chemie International Edition in English.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about Metal carbonyl — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report