From Surf Wiki (app.surf) — the open knowledge base
HIV-1 protease
Enzyme involved with peptide bond hydrolysis in retroviruses

Enzyme involved with peptide bond hydrolysis in retroviruses
| Field | Value |
|---|---|
| image | Aspartic_protease.png |
| caption | HIV-1 protease dimer in white and grey, with peptide substrate in black and active site aspartate side chains in red. () |
| EC_number | 3.4.23.16 |
| CAS_number | 144114-21-6 |
| GO_code | GO:000419 |
| Name | HIV-1 Protease (Retropepsin) |
HIV-1 protease or PR is a retroviral aspartyl protease (retropepsin), an enzyme involved with peptide bond hydrolysis in retroviruses, that is essential for the life-cycle of HIV, the retrovirus that causes AIDS. HIV-1 PR cleaves newly synthesized polyproteins (namely, Gag and Gag-Pol) at nine cleavage sites to create the mature protein components of an HIV virion, the infectious form of a virus outside of the host cell. Without effective HIV-1 PR, HIV virions remain uninfectious.
Structure
Mature HIV protease exists as a 22 kDa homodimer, with each subunit made up of 99 amino acids. A single active site lies between the identical subunits and has the characteristic Asp-Thr-Gly (Asp25, Thr26 and Gly27) catalytic triad sequence common to aspartic proteases. As HIV-1 PR can only function as a dimer, the mature protease contains two Asp25 amino acids, one from each monomer, that act in conjunction with each other as the catalytic residues. Additionally, HIV protease has two molecular "flaps" which move a distance of up to 7 Å when the enzyme becomes associated with a substrate. This can be visualized with animations of the flaps opening and closing.
Biosynthesis

Precursor
The Gag-Pol polyprotein, which contains premature coding proteins, including HIV-1 PR. PR is located between the reverse transcriptase (which is at the C-terminus of PR) and the p6pol (which is at the N-terminus of PR) of the transframe region (TFR).
In order for this precursor to become a functional protein, each monomer must associate with another HIV-1 PR monomer to form a functional catalytic active site by each contributing the Asp25 of their respective catalytic triads.
Synthesis Mechanism
When viral HIV-RNA enters the cell, it is accompanied by a reverse transcriptase, an integrase, and a mature HIV-1 PR. The reverse transcriptase converts viral RNA into DNA, facilitating the integrase's role in incorporating viral genetic information with the host cell DNA. The viral DNA can either remain dormant in the nucleus or be transcribed into mRNA and translated by the host cell into the Gag-Pol polyprotein, which would then be cleaved into individual functional proteins (including a newly synthesized HIV-1 PR) by the mature HIV-1 PR.
The HIV-1 PR precursor catalyzes its own production by facilitating its cleavage from the Gag-Pol polyprotein in a mechanism known as auto-processing. Auto-processing of HIV-1 PR is characterized by two sequential steps: (1) the intramolecular cleavage of the N-terminus at the p6pol-protease cleavage site, which serves to finalize PR processing and increase enzymatic activity with the newly formed PR-reverse transcriptase intermediate, and (2) the intermolecular cleavage of the C-terminus at the protease-reverse transcriptase cleavage site, leading to the assembly of two PR subunits into mature dimers. Dimerization of the two subunits allows for fully functional, combined active site, characterized by two Asp25 catalytic residues (one from each monomer), to form.{{multiple image
Function
HIV-1 PR serves a dual purpose. Precursor HIV-1 PR is responsible for catalyzing its own production into mature PR enzymes via PR auto-processing. Mature protease is able to hydrolyze peptide bonds on the Gag-Pol polyproteins at nine specific sites, processing the resulting subunits into mature, fully functional proteins. These cleaved proteins, including reverse transcriptase, integrase, and RNaseH, are encoded by the coding region components necessary for viral replication.
Mechanism
As an aspartic protease, the dimerized HIV-1 PR functions through the aspartyl group complex, in order to perform hydrolysis. Of the two Asp25 residues on the combined catalytic active site of HIV-1 PR, one is deprotonated while the other is protonated, due to pKa differences from the micro-environment.
In a general aspartic protease mechanism, once the substrate is properly bound to the active site of the enzyme, the deprotonated Asp25 catalytic amino acid undergoes base catalysis, rendering the incoming water molecule a better nucleophile by deprotonating it. The resulting hydroxyl ion attacks the carbonyl carbon of the peptide bond, forming an intermediate with a transient oxyanion, which is stabilized by the initially protonated Asp25. The oxyanion re-forms a double bond, leading to the cleavage of the peptide bond between the two amino acids, while the initially deprotonated Asp25 undergoes acid catalysis to donate its proton to the amino group, making the amino group a better leaving group for complete peptide bond cleavage and returning to its original deprotonated state.
While HIV-1 PR shares many of the same characteristics as a non-viral aspartic protease, some evidence has shown that HIV-1 PR catalyzes hydrolysis in a concerted manner; in other words, the nucleophilic water molecule and the protonated Asp25 simultaneously attack the scissile peptide bond during catalysis.

As a drug target
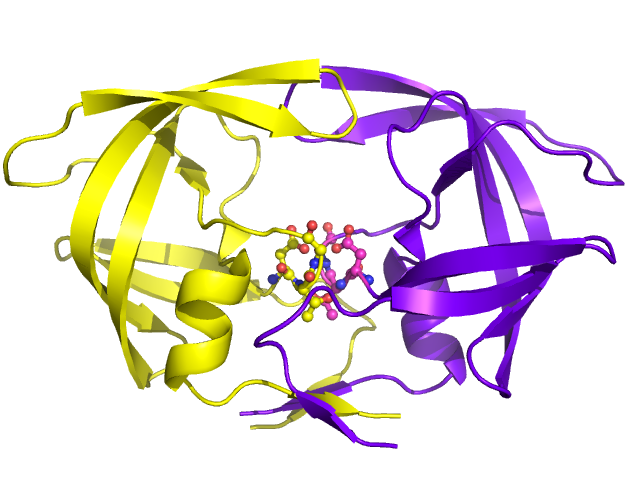
There are ten HIV-1 PR inhibitors that are currently approved by the Food and Drug Administration: indinavir, saquinavir, ritonavir, nelfinavir, lopinavir, amprenavir, fosamprenevir, atazanavir, tipranavir, and darunavir. Many of the inhibitors have different molecular components and thus different mechanistic actions, such as blocking the active site. Their functional roles also extend to influencing circulation concentrations of other inhibitor drugs (ritonavir) and being used only for certain circumstances in which the virus exhibits tolerance of other inhibitors (tipranavir).
Evolution and resistance
Due to the high mutation rates of retroviruses, especially due to mutationally sensitive regions (notably the region containing the catalytic triad sequence), and considering that changes to a few amino acids within HIV protease can render it much less visible to an inhibitor, the active site of this enzyme can change rapidly when under the selective pressure of replication-inhibiting drugs. Despite most of known resistant mutations can affect the stability of the HIV-1 PR, this protein could still perform its catalytic activity, sometimes facilitated by compensatory mutations.
Two types of mutations are generally associated with increasing drug resistance: "major" mutations and "secondary" mutations. Major mutations involve a mutation on the active site of HIV-1 PR, preventing the selective inhibitors from binding it. Secondary mutations refer to molecular changes on the periphery of the enzyme due to prolonged exposure of similar chemicals, potentially affecting inhibitor specificity for HIV-1 PR.
One approach to minimizing the development of drug-resistance in HIV is to administer a combination of drugs which inhibit several key aspects of the HIV replication cycle simultaneously, rather than one drug at a time. Other drug therapy targets include reverse transcriptase, virus attachment, membrane fusion, cDNA integration and virion assembly.
References
References
- (1990). "The structure and function of the aspartic proteinases". Annual Review of Biophysics and Biophysical Chemistry.
- (January 2003). "HIV-1 protease: mechanism and drug discovery". Organic & Biomolecular Chemistry.
- (June 2014). "The role of select subtype polymorphisms on HIV-1 protease conformational sampling and dynamics". The Journal of Biological Chemistry.
- (April 2015). "HIV protease inhibitors: a review of molecular selectivity and toxicity". HIV/AIDS: Research and Palliative Care.
- (February 1989). "Activity of purified biosynthetic proteinase of human immunodeficiency virus on natural substrates and synthetic peptides". Proceedings of the National Academy of Sciences of the United States of America.
- (July 1988). "Active human immunodeficiency virus protease is required for viral infectivity". Proceedings of the National Academy of Sciences of the United States of America.
- (April 2004). "HIV-1 protease molecular dynamics of a wild-type and of the V82F/I84V mutant: possible contributions to drug resistance and a potential new target site for drugs". Protein Science.
- (March 2005). "Folding regulates autoprocessing of HIV-1 protease precursor". The Journal of Biological Chemistry.
- (August 2004). "Initial cleavage of the human immunodeficiency virus type 1 GagPol precursor by its activated protease occurs by an intramolecular mechanism". Journal of Virology.
- (December 1989). "Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 A resolution". Science.
- (September 1999). "Autoprocessing of HIV-1 protease is tightly coupled to protein folding". Nature Structural Biology.
- (August 1994). "Kinetics and mechanism of autoprocessing of human immunodeficiency virus type 1 protease from an analog of the Gag-Pol polyprotein". Proceedings of the National Academy of Sciences of the United States of America.
- (February 1996). "A transient precursor of the HIV-1 protease. Isolation, characterization, and kinetics of maturation". The Journal of Biological Chemistry.
- (November 2008). "HIV-1 protease function and structure studies with the simplicial neighborhood analysis of protein packing method". Proteins.
- (July 2013). "Understanding HIV-1 protease autoprocessing for novel therapeutic development". [[Future Medicinal Chemistry]].
- (November 1996). "Ionization states of the catalytic residues in HIV-1 protease". Nature Structural Biology.
- (August 1996). "A combined quantum/classical molecular dynamics study of the catalytic mechanism of HIV protease". Journal of Molecular Biology.
- (February 1991). "Structure at 2.5-A resolution of chemically synthesized human immunodeficiency virus type 1 protease complexed with a hydroxyethylene-based inhibitor". Biochemistry.
- (2007). "Rang and Dale's pharmacology". Churchill Livingstone/Elsevier.
- (September 2011). "Influence of drug transport proteins on the pharmacokinetics and drug interactions of HIV protease inhibitors". Journal of Pharmaceutical Sciences.
- (February 2003). "Selection of high-level resistance to human immunodeficiency virus type 1 protease inhibitors". Antimicrobial Agents and Chemotherapy.
- (August 1989). "Complete mutagenesis of the HIV-1 protease". Nature.
- (2011-05-13). "The Evolution of Catalytic Function in the HIV-1 Protease". Journal of Molecular Biology.
- (2022-07-01). "The evolution of the HIV-1 protease folding stability". Virus Evolution.
- (December 1999). "Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy". AIDS.
- (October 2000). "New targets for inhibitors of HIV-1 replication". Nature Reviews. Molecular Cell Biology.
- (December 2007). "The design of drugs for HIV and HCV". Nature Reviews. Drug Discovery.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about HIV-1 protease — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report