From Surf Wiki (app.surf) — the open knowledge base
Crouzon syndrome
Genetic disorder of the skull and face
.jpg)
Genetic disorder of the skull and face
| Field | Value |
|---|---|
| name | Crouzon syndrome |
| image | File: Baby with Crouzon Syndrome.jpg |
| caption | Baby with Crouzon syndrome |
| field | Medical genetics |
Branchial arch syndrome
Crouzon syndrome is an autosomal dominant genetic disorder caused by a mutation in a gene on chromosome 10 that controls the body's production of fibroblast growth factor receptor 2 (FGFR2). Crouzon syndrome is named for Octave Crouzon, a French physician who first described this disorder. First called "craniofacial dysostosis" ("craniofacial" refers to the skull and face, and "dysostosis" refers to malformation of bone), the disorder was characterized by a number of clinical features which can be described by the rudimentary meanings of its former name. The developing fetus's skull and facial bones fuse early or are unable to expand. Thus, normal bone growth cannot occur. Fusion of different sutures leads to abnormal patterns of growth of the skull.
Signs and symptoms
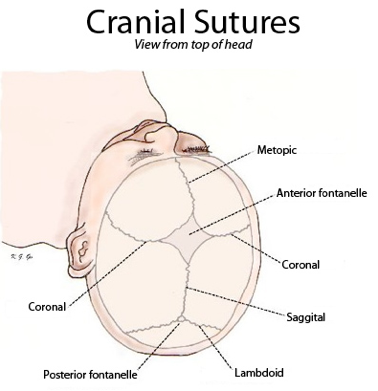
A defining characteristic of Crouzon syndrome is craniosynostosis, which results in an abnormal head shape. This is present in combinations of: frontal bossing, trigonocephaly (fusion of the metopic suture), brachycephaly (fusion of the coronal suture), dolichocephaly (fusion of the sagittal suture), plagiocephaly (unilateral premature closure of lambdoid and coronal sutures), oxycephaly (fusion of coronal and lambdoidal sutures), and complex craniosynostosis (premature closure of some or all sutures).
Exophthalmos (bulging eyes due to shallow eye sockets after early fusion of surrounding bones), hypertelorism (greater than normal distance between the eyes), and psittichorhina (beak-like nose) are also very common features. Other facial characteristics that are present in many cases include external strabismus and hypoplastic maxilla (insufficient growth of the midface), which results in relative mandibular prognathism (protruding chin) and gives the effect of the patient having a concave face.
Most symptoms are secondary to the abnormal skull structure. Approximately 30% of people with Crouzon syndrome develop hydrocephalus. Sensorineural hearing loss is present in some cases. The abnormalities in the manner in which the eyes fit in the eye sockets can cause vision problems, the most common of which is corneal exposure that can lead to visual impairment. Some people with the condition have a restricted airway and can experience severe problems breathing.
Common features are a narrow/high-arched palate, posterior bilateral crossbite, hypodontia (missing some teeth), and crowding of teeth. Due to maxillary hypoplasia, people with Crouzon syndrome generally have a considerable permanent underbite.
Causes
The current research indicates fibroblast growth factor receptors (FGFR) FGFR2 and FGFR3 as the leading factors in causing the autosomal dominant Crouzon syndrome. These two transmembrane proteins are two of four fibroblast growth factor receptors involved in osteoblast differentiation during embryonic development; mutations amongst these receptors are involved in several genetic disorders.
There are 40 known mutations, most of which are caused by a missense mutation. FGFR2 is the most commonly mutated gene, a missense at cysteine 342 in exon 9, which creates a gain-of-function. The FGFR2lllc isoform, created via alternative splicing of exon 3 of the FGFR2 gene, uses exon 9 and is used in mesenchymal stem cells to control ossification. However, the mutation constitutively activates the transmembrane protein via a disulfide bond formed incorrectly due to the loss of cysteine 342. FGFR3 is expressed more in the frontal bones during embryonic development, guiding cranial bone development. A point mutation causes constitutive activation of tyrosine in the activation loop, located in the cytosolic region of the protein, leading to accelerated differentiation of frontal osteoblasts, resulting in premature fusion of frontal cranial bones.
Diagnosis
Diagnosis of Crouzon syndrome usually can occur at birth by assessing the physical appearance of the infant. Further analysis, including radiographs, magnetic resonance imaging (MRI) scans, genetic testing and CT scans can be used to confirm the diagnosis of Crouzon syndrome.
Treatment

Surgery is typically used to prevent the closure of sutures of the skull from damaging the brain's development. Without surgery, blindness and intellectual disability are typical outcomes. Without treatment, Crouzon syndrome can cause hearing and vision loss, exposure keratitis or conjunctivitis, drying of the cornea, hydrocephalus, sleep apnea, and breathing problems. To move the orbits forward, surgeons expose the skull and orbits and reshape the bone. To treat the midface deficiency, surgeons can move the lower orbit and midface bones forward. Additionally, surgery can be performed to relieve pressure inside the skull, fix a cleft lip or palate, correct a malformed jaw, straighten crooked teeth, or correct eye problems.
People with Crouzon syndrome tend to have multiple sutures involved, most specifically bilateral coronal craniosynostoses, and either open vault surgery or strip craniectomy (if the child is under 6 months) can be performed. In the latter scenario, a helmet is worn for several months following surgery.
Once treated for the cranial vault abnormalities, Crouzon patients generally go on to live a normal lifespan.
Epidemiology
Incidence of Crouzon syndrome is currently estimated at 1.6 out of every 100,000 people. It is the most common craniostenosis syndrome.
History
Crouzon syndrome was first described by Octave Crouzon in 1912. He noted the affected patients were a mother and her daughter, implying a genetic basis.
References
References
- {{WhoNamedIt. synd. 1383
- L. E. O. Crouzon. Dysostose cranio-faciale héréditaire. Bulletin de la Société des Médecins des Hôpitaux de Paris, 1912, 33: 545-555.
- "Crouzon syndrome". Genetic and Rare Diseases Information Center (GARD), [[National Institutes of Health]].
- (9 May 2005). "Ophthalmic Sequelae of Crouzon Syndrome". Ophthalmology.
- "Crouzon Syndrome". [[National Organization for Rare Disorders]] (NORD).
- (2015). "Cummings Otolaryngology". Elsevier.
- (February 2010). "Analysis of a gain-of-function FGFR2 Crouzon mutation provides evidence of loss of function activity in the etiology of cleft palate". Proc. Natl. Acad. Sci. U.S.A..
- (August 2014). "Apparently synonymous substitutions in FGFR2 affect splicing and result in mild Crouzon syndrome". BMC Med. Genet..
- (June 2014). "FGFR3 mutation causes abnormal membranous ossification in achondroplasia". Hum. Mol. Genet..
- (October 20, 1998). "FGFR-Related Craniosynostosis Syndromes.". GeneReviews.
- "Crouzon syndrome". US National Library of Medicine, National Institutes of Health.
- (2018). "Plastic Surgery: Volume 3: Craniofacial, Head and Neck Surgery and Pediatric Plastic Surgery". Elsevier.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about Crouzon syndrome — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report