From Surf Wiki (app.surf) — the open knowledge base
Cancer immunotherapy
Artificial stimulation of the immune system to treat cancer
Artificial stimulation of the immune system to treat cancer
| Field | Value |
|---|---|
| name | Cancer immunotherapy |
| image | Peptide bound to Rituximab FAB.png |
| caption | Peptide epitope of CD20 bound to rituximab's FAB |
| specialty | Tumor immunology |
Cancer immunotherapy (immuno-oncotherapy) is the stimulation of the immune system to treat cancer, improving the immune system's natural ability to fight the disease. It is an application of the fundamental research of cancer immunology (immuno-oncology) and a growing subspecialty of oncology.
Cancer immunotherapy exploits the fact that cancer cells often have tumor antigens, molecules on their surface that can bind to antibody proteins or T-cell receptors, triggering an immune system response. The tumor antigens are often proteins or other macromolecules (e.g., carbohydrates). Normal antibodies bind to external pathogens, but the modified immunotherapy antibodies bind to the tumor antigens marking and identifying the cancer cells for the immune system to inhibit or kill. The clinical success of cancer immunotherapy is highly variable between different forms of cancer; for instance, certain subtypes of gastric cancer react well to the approach whereas immunotherapy is not effective for other subtypes.
Major types of cancer immunotherapy include immune checkpoint inhibitors, which block inhibitory pathways such as PD-1/PD-L1 and CTLA-4 to enhance T cell activity against tumors. These therapies have shown effectiveness in treating cancers such as melanoma and lung cancer.
Adoptive cell therapies, including chimeric antigen receptor (CAR) T cell therapy, involve modifying a patient's immune cells to recognize cancer-specific antigens. These therapies have been particularly effective in certain blood cancers. Natural killer cell (NK) therapies and CAR-NK cell approaches are also being explored, leveraging NK cells' innate ability to target tumor cells. Other strategies include cancer vaccines, which aim to provoke an immune response against tumor-associated antigens, and may be either preventive or therapeutic. Immunomodulatory agents such as cytokines (e.g., interleukin-2, interferon-alpha) and Bacillus Calmette-Guerin (BCG) are used to enhance immune activity or alter the tumor microenvironment. Oncolytic virus therapies, which employ engineered viruses to selectively kill cancer cells while promoting systemic immunity, are also under investigation.
In 2018, American immunologist James P. Allison and Japanese immunologist Tasuku Honjo received the Nobel Prize in Physiology or Medicine for their discovery of cancer therapy by inhibition of negative immune regulation.
History
"During the 17th and 18th centuries, various forms of immunotherapy in cancer became widespread... In the 18th and 19th centuries, septic dressings enclosing ulcerative tumours were used for the treatment of cancer. Surgical wounds were left open to facilitate the development of infection, and purulent sores were created deliberately... One of the most well-known effects of microorganisms on ... cancer was reported in 1891, when an American surgeon, William Coley, inoculated patients having inoperable tumours with [Streptococcus pyogenes ]." "Coley [had] thoroughly reviewed the literature available at that time and found 38 reports of cancer patients with accidental or iatrogenic feverish erysipelas. In 12 patients, the sarcoma or carcinoma had completely disappeared; the others had substantially improved. Coley decided to attempt the therapeutic use of iatrogenic erysipelas..." "Coley developed a toxin that contained heat-killed bacteria Streptococcus pyogenes and [Serratia marcescens ]. Until 1963, this treatment was used for the treatment of sarcoma." 51.9% of [Coley's] patients with inoperable soft-tissue sarcomas showed complete tumour regression and survived for more than 5 years, and 21.2% of the patients had no clinical evidence of tumour at least 20 years after this treatment..."
In the 1980's, researchers at the National Cancer Institute's Center for Cancer Research (CCR) began exploring the then-heretical idea that a patient's immune system could be harnessed to fight cancer. These researchers included Michael Potter, Ira Pastan, and Steven Rosenberg who developed approaches including monoclonal antibody-based immunotoxins, checkpoint blockade drugs, cytokine-based therapies, and adoptive cell therapy studies.
Types and categories
There are several types of immunotherapy used to treat cancer:
- Immune checkpoint inhibitors: drugs that block immune system checkpoints to allow immune cells to respond more strongly to the cancer.
- T-cell transfer therapy: a treatment that takes T-cells from the tumor and selects or changes them in the lab to better attack cancer cells, then reintroduces them into the patient.
- Monoclonal antibodies: designed to bind to specific targets on cancer cells, marking cancer cells so that they will be better seen and destroyed by the immune system.
- Treatment vaccines: also known as therapeutic cancer vaccines, help the immune system learn to recognize and react to mutant proteins specific to the tumor and destroy cancer cells containing them.
- Immune system modulators: agents that enhance the body's immune response against cancer.
Immunotherapies can be categorized as active or passive based on their ability to engage the host immune system against cancer. Active immunotherapy specifically targets tumor cells via the immune system. Examples include therapeutic cancer vaccines (also known as treatment vaccines, which are designed to boost the body's immune system to fight cancer), CAR-T cells, and targeted antibody therapies. In contrast, passive immunotherapy does not directly target tumor cells, but enhances the ability of the immune system to attack cancer cells. Examples include checkpoint inhibitors and cytokines.
Active cellular therapies aim to destroy cancer cells by recognition of distinct markers known as antigens. In cancer vaccines, the goal is to generate an immune response to these antigens through a vaccine. Currently, only one vaccine (sipuleucel-T for prostate cancer) has been approved. In cell-mediated therapies like CAR-T cell therapy, immune cells are extracted from the patient, genetically engineered to recognize tumor-specific antigens, and returned to the patient. Cell types that can be used in this way are natural killer (NK) cells, lymphokine-activated killer cells, cytotoxic T cells, and dendritic cells. Finally, specific antibodies can be developed that recognize cancer cells and target them for destruction by the immune system. Examples of such antibodies include rituximab (targeting CD-20), trastuzumab (targeting HER-2), and cetuximab (targeting EGFR).
Passive antibody therapies aim to increase the activity of the immune system without specifically targeting cancer cells. For example, cytokines directly stimulate the immune system and increase immune activity. Checkpoint inhibitors target proteins (immune checkpoints) that normally dampen the immune response. This enhances the ability of the immune system to attack cancer cells. Current research is identifying new potential targets to enhance immune function. Approved checkpoint inhibitors include antibodies such as ipilimumab, nivolumab, and pembrolizumab.
Cellular immunotherapy
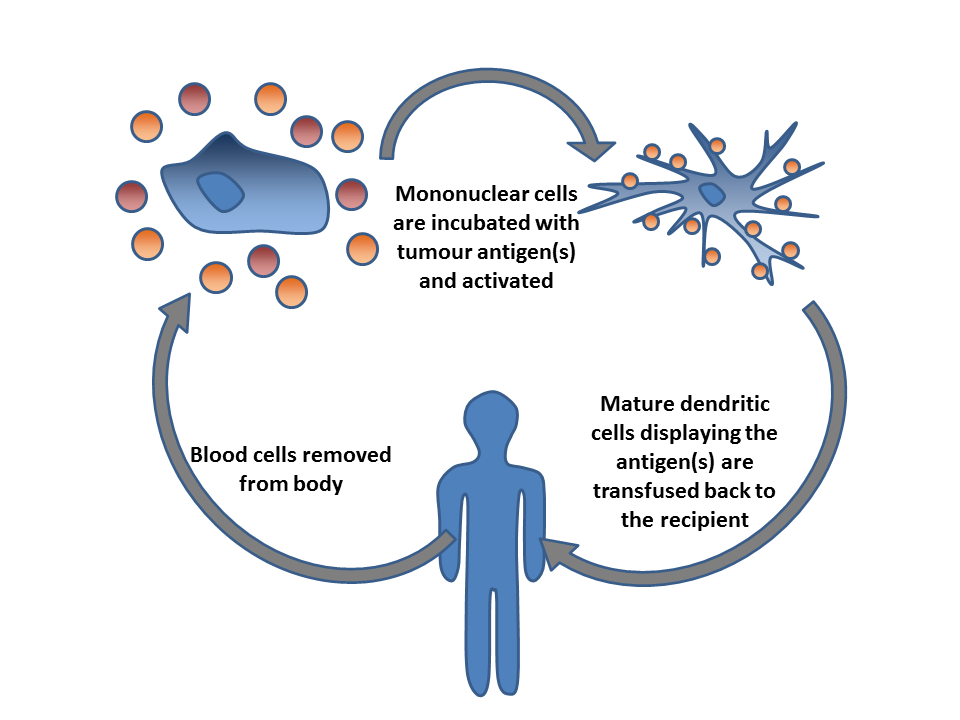
Dendritic cell therapy
Dendritic cell therapy provokes anti-tumor responses by causing dendritic cells to present tumor antigens to lymphocytes, which activates them, priming them to kill other cells that present the antigen. Dendritic cells are antigen-presenting cells (APCs) in the mammalian immune system. In cancer treatment, they aid cancer antigen targeting. The only approved cellular cancer therapy based on dendritic cells is sipuleucel-T.
One method of inducing dendritic cells to present tumor antigens is by vaccination with autologous tumor lysates or short peptides (small parts of the protein that correspond to the protein antigens on cancer cells). These peptides are often given in combination with adjuvants (highly immunogenic substances) to increase the immune and anti-tumor responses. Other adjuvants include proteins or other chemicals that attract and/or activate dendritic cells, such as granulocyte-macrophage colony-stimulating factor (GM-CSF). The most common sources of antigens used for dendritic cell vaccine in glioblastoma (GBM) as an aggressive brain tumor were whole tumor lysate, CMV antigen RNA and tumor-associated peptides like EGFRvIII.
Dendritic cells can also be activated in vivo by making tumor cells express GM-CSF. This can be achieved by either genetically engineering tumor cells to produce GM-CSF or by infecting tumor cells with an oncolytic virus that expresses GM-CSF.
Another strategy is to remove dendritic cells from the blood of a patient and activate them outside the body. The dendritic cells are activated in the presence of tumor antigens, which may be a single tumor-specific peptide/protein or a tumor cell lysate (a solution of broken-down tumor cells). These cells (with optional adjuvants) are infused and provoke an immune response.
Dendritic cell therapies include the use of antibodies that bind to receptors on the surface of dendritic cells. Antigens can be added to the antibody and can induce the dendritic cells to mature and provide immunity to the tumor. Dendritic cell receptors such as TLR3, TLR7, TLR8 or CD40 have been used as antibody targets. Dendritic cell-NK cell interface also has an important role in immunotherapy. The design of new dendritic cell-based vaccination strategies should also encompass NK cell-stimulating potency. It is critical to systematically incorporate NK cells monitoring as an outcome in antitumor DC-based clinical trials.
Drugs
Sipuleucel-T (Provenge) was approved for treatment of asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer in 2010. The treatment consists of removal of antigen-presenting cells from blood by leukapheresis and growing them with the fusion protein PA2024 made from GM-CSF and prostate-specific prostatic acid phosphatase (PAP) and reinfused. This process is repeated three times.
Adoptive T-cell therapy

Adoptive T cell therapy is a form of passive immunization by the transfusion of T-cells. They are found in blood and tissue and typically activate when they find foreign pathogens. Activation occurs when the T-cell's surface receptors encounter cells that display parts of foreign proteins (either on their surface or intracellularly). These can be either infected cells or other antigen-presenting cells (APCs). The latter are found in normal tissue and in tumor tissue, where they are known as tumor-infiltrating lymphocytes (TILs). They are activated by the presence of APCs such as dendritic cells that present tumor antigens. Although these cells can attack tumors, the tumor microenvironment is highly immunosuppressive, interfering with immune-mediated tumour death.
Multiple ways of producing tumour-destroying T-cells have been developed. Most commonly, T-cells specific to a tumor antigen can be removed from a tumor sample (TILs) or filtered from blood. The T-cells can optionally be modified in various ways, cultured and infused into patients. T cells can be modified via genetic engineering, producing CAR-T cell or TCR T cells or by exposing the T cells to tumor antigens in a non-immunosuppressive environment, that they recognize as foreign and learn to attack.
Another approach is transfer of haploidentical γδ T cells or natural killer cells from a healthy donor. The major advantage of this approach is that these cells do not cause graft-versus-host disease. The disadvantage is that transferred cells frequently have impaired function.
Tumor-derived T cell therapy
The simplest example involves removing TILs from a tumor, culturing but not modifying them, and infusing the result back into the tumour. The first therapy of this type, Lifileucel, achieved US Food and Drug Administration (FDA) approval in February 2024.
CAR-T cell therapy
Main article: Chimeric antigen receptor T cell
The premise of CAR-T immunotherapy is to modify T cells to recognize cancer cells in order to target and destroy them. Scientists harvest T cells from people, genetically alter them to add a chimeric antigen receptor (CAR) that specifically recognizes cancer cells, then infuse the resulting CAR-T cells into patients to attack their tumors.
Tisagenlecleucel (Kymriah), a chimeric antigen receptor (CAR-T) therapy, was approved by the FDA in 2017 to treat acute lymphoblastic leukemia (ALL). This treatment removes CD19 positive cells (B-cells) from the body (including the diseased cells, but also normal antibody-producing cells).
Axicabtagene ciloleucel (Yescarta) is another CAR-T therapeutic, approved in 2017 for treatment of diffuse large B-cell lymphoma (DLBCL).
Multifunctional alginate scaffolds
Multifunctional alginate scaffolds for T cell engineering and release (MASTER) is a technique for in situ engineering, replication and release of genetically engineered T cells. It is an evolution of CAR T cell therapy. T cells are extracted from the patient and mixed with a genetically engineered virus that contains a cancer-targeting gene (as with CAR T). The mixture is then added to a MASTER (scaffold), which absorbs them. The MASTER contains antibodies that activate the T cells and interleukins that trigger cell proliferation. The MASTER is then implanted into the patient. The activated T cells interact with the viruses to become CAR T cells. The interleukins stimulate these CAR T cells to proliferate, and the CAR T cells exit the MASTER to attack the cancer. The technique takes hours instead of weeks. And because the cells are younger, they last longer in the body, show stronger potency against cancer, and display fewer markers of exhaustion. These features were demonstrated in mouse models. The treatment was more effective and longer-lasting against lymphoma.
T cell receptor T cell therapy
Antibody therapy

Antibody types
Conjugation
Two types are used in cancer treatments:
- Naked monoclonal antibodies are antibodies without added elements. Most antibody therapies use this antibody type.
- Conjugated monoclonal antibodies are joined to another molecule, which is either cytotoxic or radioactive. The toxic chemicals are those typically used as chemotherapy drugs, but other toxins can be used. The antibody binds to specific antigens on cancer cell surfaces, directing the therapy to the tumor. Radioactive compound-linked antibodies are referred to as radiolabelled. Chemolabelled or immunotoxins antibodies are tagged with chemotherapeutic molecules or toxins, respectively. Research has also demonstrated conjugation of a TLR agonist to an anti-tumor monoclonal antibody.
Fc regions
Fc's ability to bind Fc receptors is important because it allows antibodies to activate the immune system. Fc regions are varied: they exist in numerous subtypes and can be further modified, for example with the addition of sugars in a process called glycosylation. Changes in the Fc region can alter an antibody's ability to engage Fc receptors and, by extension, will determine the type of immune response that the antibody triggers. For example, immune checkpoint blockers targeting PD-1 are antibodies designed to bind PD-1 expressed by T cells and reactivate these cells to eliminate tumors. Anti-PD-1 drugs contain not only a Fab region that binds PD-1 but also an Fc region. Experimental work indicates that the Fc portion of cancer immunotherapy drugs can affect the outcome of treatment. For example, anti-PD-1 drugs with Fc regions that bind inhibitory Fc receptors can have decreased therapeutic efficacy. Imaging studies have further shown that the Fc region of anti-PD-1 drugs can bind Fc receptors expressed by tumor-associated macrophages. This process removes the drugs from their intended targets (i.e. PD-1 molecules expressed on the surface of T cells) and limits therapeutic efficacy. Furthermore, antibodies targeting the co-stimulatory protein CD40 require engagement with selective Fc receptors for optimal therapeutic efficacy. Together, these studies underscore the importance of Fc status in antibody-based immune checkpoint targeting strategies.
Human/non-human antibodies
Antibodies can come from a variety of sources, including human cells, mice, and a combination of the two (chimeric antibodies). Different sources of antibodies can provoke different kinds of immune responses. For example, the human immune system can recognize mouse antibodies (also known as murine antibodies) and trigger an immune response against them. This could reduce the effectiveness of the antibodies as a treatment and cause an immune reaction. Chimeric antibodies attempt to reduce murine antibodies' immunogenicity by replacing part of the antibody with the corresponding human counterpart. Humanized antibodies are almost completely human; only the complementarity determining regions of the variable regions are derived from murine sources. Human antibodies have been produced using unmodified human DNA.
Mechanism of action
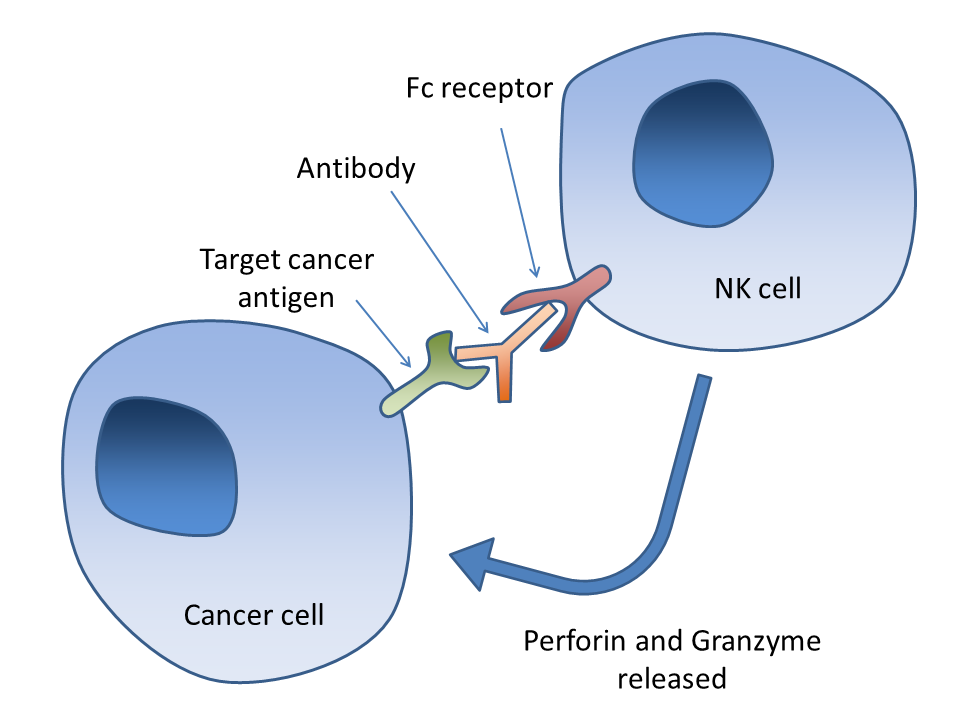
Antibody-dependent cell-mediated cytotoxicity (ADCC)
Antibody-dependent cell-mediated cytotoxicity (ADCC) requires antibodies to bind to target cell surfaces. Antibodies are formed of a binding region (Fab) and the Fc region that can be detected by immune system cells via their Fc surface receptors. Fc receptors are found on many immune system cells, including NK cells. When NK cells encounter antibody-coated cells, the latter's Fc regions interact with their Fc receptors, releasing perforin and granzyme B to kill the tumor cell. Examples include rituximab, ofatumumab, elotuzumab, and alemtuzumab. Antibodies under development have altered Fc regions that have higher affinity for a specific type of Fc receptor, FcγRIIIA, which can dramatically increase effectiveness.
Anti-CD47 therapy
Many tumor cells overexpress CD47 to escape immunosurveilance of host immune system. CD47 binds to its receptor signal-regulatory protein alpha (SIRPα) and downregulate phagocytosis of tumor cell. Therefore, anti-CD47 therapy aims to restore clearance of tumor cells. Additionally, growing evidence supports the employment of tumor antigen-specific T cell response in response to anti-CD47 therapy. A number of therapeutics are being developed, including anti-CD47 antibodies, engineered decoy receptors, anti-SIRPα antibodies and bispecific agents. As of 2017, wide range of solid and hematologic malignancies were being clinically tested.
Anti-GD2 antibodies
Carbohydrate antigens on the surface of cells can be used as targets for immunotherapy. GD2 is a ganglioside found on the surface of many types of cancer cell including neuroblastoma, retinoblastoma, melanoma, small cell lung cancer, brain tumors, osteosarcoma, rhabdomyosarcoma, Ewing's sarcoma, liposarcoma, fibrosarcoma, leiomyosarcoma and other soft tissue sarcomas. It is not usually expressed on the surface of normal tissues, making it a good target for immunotherapy. As of 2014, clinical trials were underway.
Complement Activation
The complement system includes blood proteins that can cause cell death after an antibody binds to the cell surface (the classical complement pathway, among the ways of complement activation). Generally, the system deals with foreign pathogens but can be activated with therapeutic antibodies in cancer. The system can be triggered if the antibody is chimeric, humanized, or human; as long as it contains the IgG1 Fc region. Complement can lead to cell death by activation of the membrane attack complex, known as complement-dependent cytotoxicity; enhancement of antibody-dependent cell-mediated cytotoxicity; and CR3-dependent cellular cytotoxicity. Complement-dependent cytotoxicity occurs when antibodies bind to the cancer cell surface, the C1 complex binds to these antibodies and subsequently, protein pores are formed in cancer cell membrane.
Blocking
Antibody therapies can also function by binding to proteins and physically blocking them from interacting with other proteins. Checkpoint inhibitors (CTLA-4, PD-1, and PD-L1) operate by this mechanism. Briefly, checkpoint inhibitors are proteins that normally help to slow immune responses and prevent the immune system from attacking normal cells. Checkpoint inhibitors bind these proteins and prevent them from functioning normally, which increases the activity of the immune system. Examples include durvalumab, ipilimumab, nivolumab, and pembrolizumab.
FDA-approved antibodies
| Antibody | Brand name | Type | Target | Approval date | Approved treatment(s) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alemtuzumab | Campath | humanized | CD52 | 2001 | B-cell chronic lymphocytic leukemia (CLL) | ||||||
| Atezolizumab | Tecentriq | humanized | PD-L1 | 2016 | url=https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm501762.htm | archive-url=https://web.archive.org/web/20160519114208/http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm501762.htm | archive-date=19 May 2016 | title=FDA approves new, targeted treatment for bladder cancer | date=18 May 2016 | publisher=FDA | access-date=20 May 2016}} |
| Atezolizumab/hyaluronidase | Tecentriq Hybreza | humanized | PD-L1 | 2024 | title=FDA approves atezolizumab and hyaluronidase-tqjs | website=U.S. Food and Drug Administration | date=12 September 2024 | url=https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-and-hyaluronidase-tqjs-subcutaneous-injection | access-date=14 September 2024 | archive-date=14 September 2024 | archive-url=https://web.archive.org/web/20240914055712/https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-and-hyaluronidase-tqjs-subcutaneous-injection }} |
| Avelumab | Bavencio | human | PD-L1 | 2017 | metastatic Merkel cell carcinoma | ||||||
| Durvalumab | Imfinzi | human | PD-L1 | 2017 | bladder cancer non-small cell lung cancer | ||||||
| Elotuzumab | Empliciti | humanized | SLAMF7 | 2015 | multiple myeloma | ||||||
| Ipilimumab | Yervoy | human | CTLA4 | 2011 | metastatic melanoma | ||||||
| Nivolumab | Opdivo | human | PD-1 | 2014 | unresectable or metastatic melanoma, squamous non-small cell lung cancer, Renal cell carcinoma, colorectal cancer, hepatocellular carcinoma, classical hodgkin lymphoma | ||||||
| Ofatumumab | Arzerra | human | CD20 | 2009 | refractory CLL | ||||||
| Pembrolizumab | Keytruda | humanized | PD-1 | 2014 | unresectable or metastatic melanoma, squamous non-small cell lung cancer (NSCLC), Hodgkin's lymphoma, Merkel-cell carcinoma (MCC), primary mediastinal B-cell lymphoma (PMBCL), stomach cancer, cervical cancer | ||||||
| Rituximab | Rituxan, Mabthera | chimeric | CD20 | 1997 | non-Hodgkin lymphoma | ||||||
| Rituximab/hyaluronidase | Rituxan Hycela | chimeric | CD20 | 2017 | follicular lymphoma, diffuse large B-cell lymphoma, chronic lymphocytic leukemia | ||||||
| Trastuzumab | Rituxan Hycela | humanized | HER2/neu | 1998 | breast cancer, gastric or gastroesophageal junction adenocarcinoma |
Alemtuzumab
Alemtuzumab (Campath-1H) is an anti-CD52 humanized IgG1 monoclonal antibody indicated for the treatment of fludarabine-refractory chronic lymphocytic leukemia (CLL), cutaneous T-cell lymphoma, peripheral T-cell lymphoma and T-cell prolymphocytic leukemia. CD52 is found on 95% of peripheral blood lymphocytes (both T-cells and B-cells) and monocytes, but its function in lymphocytes is unknown. It binds to CD52 and initiates its cytotoxic effect by complement fixation and ADCC mechanisms. Due to the antibody target (cells of the immune system), common complications of alemtuzumab therapy are infection, toxicity and myelosuppression.
Atezolizumab
Atezolizumab/hyaluronidase
Avelumab
Durvalumab
Main article: Durvalumab
Durvalumab (Imfinzi) is a human immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that blocks the interaction of programmed cell death ligand 1 (PD-L1) with the PD-1 and CD80 (B7.1) molecules. Durvalumab is approved for the treatment of patients with locally advanced or metastatic urothelial carcinoma who:
- have disease progression during or following platinum-containing chemotherapy.
- have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. On 16 February 2018, the Food and Drug Administration approved durvalumab for patients with unresectable stage III non-small cell lung cancer (NSCLC) whose disease has not progressed following concurrent platinum-based chemotherapy and radiation therapy.
Elotuzumab
Ipilimumab
Ipilimumab (Yervoy) is a human IgG1 antibody that binds the surface protein CTLA4. In normal physiology T-cells are activated by two signals: the T-cell receptor binding to an antigen-MHC complex and T-cell surface receptor CD28 binding to CD80 or CD86 proteins. CTLA4 binds to CD80 or CD86, preventing the binding of CD28 to these surface proteins and therefore negatively regulates the activation of T-cells.
Active cytotoxic T-cells are required for the immune system to attack melanoma cells. Normally inhibited active melanoma-specific cytotoxic T-cells can produce an effective anti-tumor response. Ipilimumab can cause a shift in the ratio of regulatory T-cells to cytotoxic T-cells to increase the anti-tumor response. Regulatory T-cells inhibit other T-cells, which may benefit the tumor.
Nivolumab
Main article: [Nivolumab}}[[Nivolumab]] is a human [[IgG4]] antibody that prevents T-cell inactivation by blocking the binding of [PD-L1, [programmed cell death 1 ligand 1]] or programmed cell death 1 ligand 2 (PD-L1 or PD-L2), a protein expressed by cancer cells, with [Programmed cell death protein 1
Ofatumumab
Ofatumumab is a second generation human IgG1 antibody that binds to CD20. It is used in the treatment of chronic lymphocytic leukemia (CLL) because the cancerous cells of CLL are usually CD20-expressing B-cells. Unlike rituximab, which binds to a large loop of the CD20 protein, ofatumumab binds to a separate, small loop. This may explain their different characteristics. Compared to rituximab, ofatumumab induces complement-dependent cytotoxicity at a lower dose with less immunogenicity.
Pembrolizumab
As of 2019, pembrolizumab, which blocks PD-1, programmed cell death protein 1, has been used via intravenous infusion to treat inoperable or metastatic melanoma, metastatic non-small cell lung cancer (NSCLC) in certain situations, as a second-line treatment for head and neck squamous cell carcinoma (HNSCC), after platinum-based chemotherapy, and for the treatment of adult and pediatric patients with refractory classic Hodgkin's lymphoma (cHL). It is also indicated for certain patients with urothelial carcinoma, stomach cancer and cervical cancer.
Rituximab
Rituximab is a chimeric monoclonal IgG1 antibody specific for CD20, developed from its parent antibody Ibritumomab. As with ibritumomab, rituximab targets CD20, making it effective in treating certain B-cell malignancies. These include aggressive and indolent lymphomas such as diffuse large B-cell lymphoma and follicular lymphoma and leukemias such as B-cell chronic lymphocytic leukemia. Although the function of CD20 is relatively unknown, CD20 may be a calcium channel involved in B-cell activation. The antibody's mode of action is primarily through the induction of ADCC and complement-mediated cytotoxicity. Other mechanisms include apoptosis and cellular growth arrest. Rituximab also increases the sensitivity of cancerous B-cells to chemotherapy.
Trastuzumab
{{anchor|Immune checkpoint blockade}} Immune checkpoint blockade ==
Main article: Checkpoint inhibitor, Immunotherapy

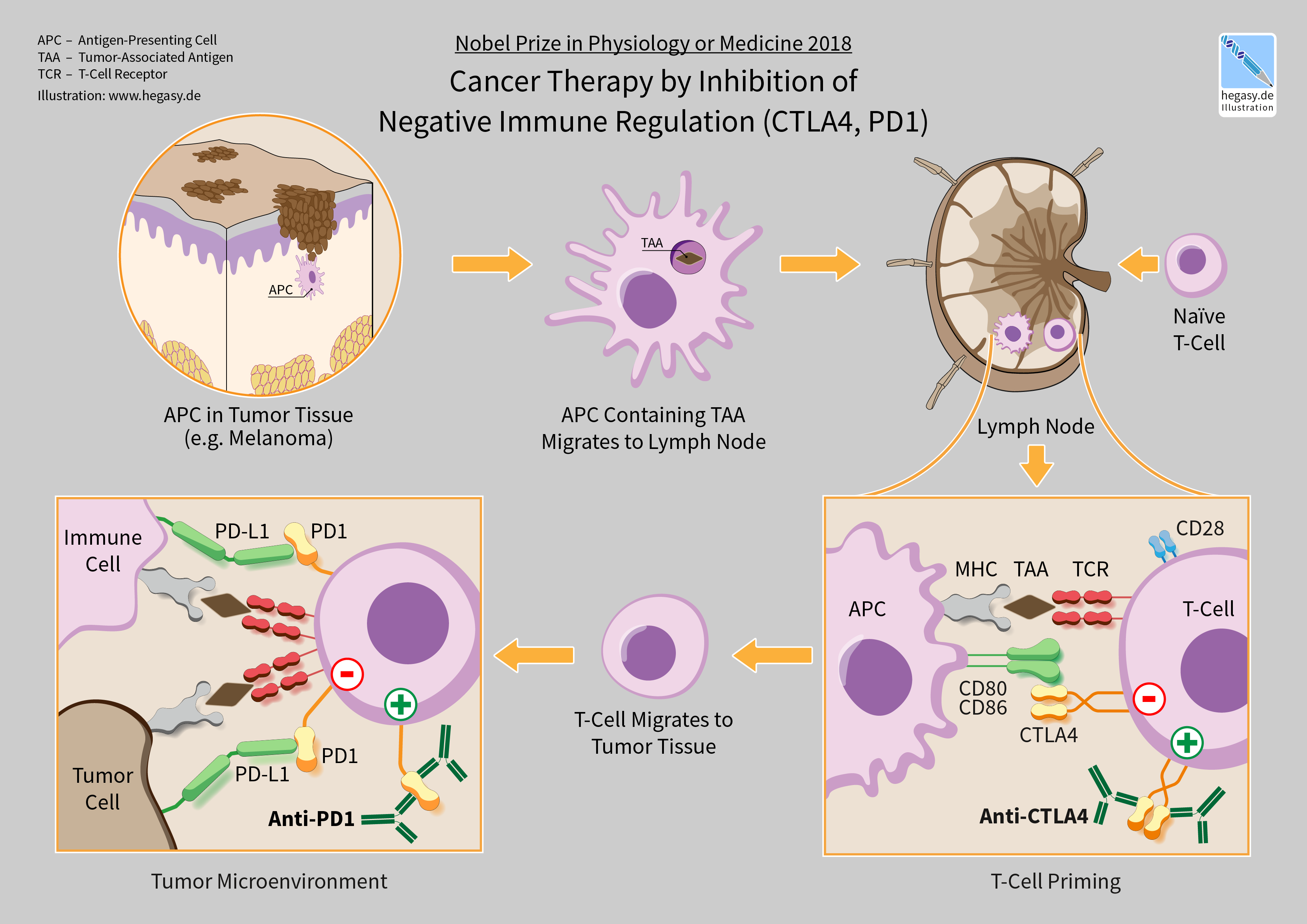
Immune checkpoints affect the immune system function. Immune checkpoints can be stimulatory or inhibitory. Tumors can use these checkpoints to protect themselves from immune system attacks. Checkpoint therapies approved as of 2012 block inhibitory checkpoint receptors. Blockade of negative feedback signaling to immune cells thus results in an enhanced immune response against tumors. As of 2020, immune checkpoint blockade therapies have varied effectiveness. In Hodgkin lymphoma and natural killer T-cell lymphoma, response rates are high, at 50–60%. Response rates are quite low for breast and prostate cancers, however. A major challenge are the large variations in responses to immunocheckpoint inhibitors, some patients showing spectacular clinical responses while no positive effects are seen in others. A plethora of possible reasons for the absence of efficacy in many patients have been proposed, but the biomedical community has still to begin to find consensus in this respect. For instance, a recent paper documented that infection with Helicobacter pylori would negatively influence the effects of immunocheckpoint inhibitors in gastric cancer., but this notion was quickly challenged by others.
One ligand-receptor interaction under investigation is the interaction between the transmembrane programmed cell death 1 protein (PDCD1, PD-1; also known as CD279) and its ligand, PD-1 ligand 1 (PD-L1, CD274). PD-L1 on the cell surface binds to PD1 on an immune cell surface, which inhibits immune cell activity. Among PD-L1 functions is a key regulatory role on T cell activities. It appears that (cancer-mediated) upregulation of PD-L1 on the cell surface may inhibit T cells that might otherwise attack. PD-L1 on cancer cells also inhibits FAS- and interferon-dependent apoptosis, protecting cells from cytotoxic molecules produced by T cells. Antibodies that bind to either PD-1 or PD-L1 and therefore block the interaction may allow the T-cells to attack the tumor.
CTLA-4 blockade
The first checkpoint antibody approved by the FDA was ipilimumab, approved in 2011 to treat melanoma. It blocks the immune checkpoint molecule CTLA-4. As of 2012, clinical trials have also shown some benefits of anti-CTLA-4 therapy on lung cancer or pancreatic cancer, specifically in combination with other drugs. In on-going trials the combination of CTLA-4 blockade with PD-1 or PD-L1 inhibitors is tested on different types of cancer.
However, as of 2015 it is known that patients treated with checkpoint blockade (specifically CTLA-4 blocking antibodies), or a combination of check-point blocking antibodies, are at high risk of having immune-related adverse events such as dermatologic, gastrointestinal, endocrine, or hepatic autoimmune reactions. These are most likely due to the breadth of the induced T-cell activation when anti-CTLA-4 antibodies are administered by injection in the bloodstream.
A 2024 cohort study of ICI use during pregnancy showed no overreporting of specific adverse effects on pregnancy, fetal, and/or newborn outcomes, interestingly.
Using a mouse model of bladder cancer, researchers have found that a local injection of a low dose anti-CTLA-4 in the tumour area had the same tumour inhibiting capacity as when the antibody was delivered in the blood. At the same time the levels of circulating antibodies were lower, suggesting that local administration of the anti-CTLA-4 therapy might result in fewer adverse events.
PD-1 inhibitors
Main article: PD-1 and PD-L1 inhibitors
Initial clinical trial results with IgG4 PD1 antibody nivolumab were published in 2010. It was approved in 2014. Nivolumab is approved to treat melanoma, lung cancer, kidney cancer, bladder cancer, head and neck cancer, and Hodgkin's lymphoma. A 2016 clinical trial for non-small cell lung cancer failed to meet its primary endpoint for treatment in the first-line setting, but is FDA-approved in subsequent lines of therapy.
Pembrolizumab (Keytruda) is another PD1 inhibitor that was approved by the FDA in 2014. Pembrolizumab is approved to treat melanoma and lung cancer.
Antibody BGB-A317 is a PD-1 inhibitor (designed to not bind Fc gamma receptor I) in early clinical trials.
PD-L1 inhibitors
Main article: PD-1 and PD-L1 inhibitors
In May 2016, PD-L1 inhibitor atezolizumab was approved for treating bladder cancer.
Anti-PD-L1 antibodies currently in development include avelumab and durvalumab, in addition to an inhibitory affimer.
CISH
Combinations
Many cancer patients do not respond to immune checkpoint blockade. Response rate may be improved by combining that with additional therapies, including those that stimulate T cell infiltration. For example, targeted therapies such as radiotherapy, vasculature targeting agents, and immunogenic chemotherapy can improve immune checkpoint blockade response in animal models.
Combining immunotherapies such as PD1 and CTLA4 inhibitors can create to durable responses.
Combinatorial ablation and immunotherapy enhances the immunostimulating response and has synergistic effects for metastatic cancer treatment.
Combining checkpoint immunotherapies with pharmaceutical agents has the potential to improve response, and as of 2018 were a target of clinical investigation. Immunostimulatory drugs such as CSF-1R inhibitors and TLR agonists have been effective.
Two independent 2024 clinical trials reported that combinations of JAK inhibitors with anti–PD-1 immunotherapy could improve efficacy. A phase 2 trial investigated the combination as a first-line therapy for metastatic non-small-cell lung cancer. Administration of itacitinib after treatment with pembrolizumab improved therapeutic response. A separate phase 1/2 trial with patients with relapsed/refractory Hodgkin's lymphoma combined ruxolitinib and nivolumab, yielding improved clinical efficacy in patients who had previously failed checkpoint blockade immunotherapy.
Cytokine therapy
Cytokines are proteins produced by many types of cells present within a tumor. They can modulate immune responses. The tumor often employs them to allow it to grow and reduce the immune response. These immune-modulating effects allow them to be used as drugs to provoke an immune response. Two commonly used cytokines are interferons and interleukins.
Interleukin-2 and interferon-α are cytokines, proteins that regulate and coordinate the behavior of the immune system. They have the ability to enhance anti-tumor activity and thus can be used as passive cancer treatments. Interferon-α is used in the treatment of hairy-cell leukaemia, AIDS-related Kaposi's sarcoma, follicular lymphoma, chronic myeloid leukaemia and melanoma. Interleukin-2 is used in the treatment of melanoma and renal cell carcinoma.
Interferon
Interferons are produced by the immune system. They are usually involved in anti-viral response, but also have use for cancer. They fall in three groups: type I (IFNα and IFNβ), type II (IFNγ) and type III (IFNλ). IFNα has been approved for use in hairy-cell leukaemia, AIDS-related Kaposi's sarcoma, follicular lymphoma, chronic myeloid leukaemia and melanoma. Type I and II IFNs have been researched extensively and although both types promote anti-tumor immune system effects, only type I IFNs have been shown to be clinically effective. IFNλ shows promise for its anti-tumor effects in animal models.
Unlike type I IFNs, Interferon gamma is not approved yet for the treatment of any cancer. However, improved survival was observed when Interferon gamma was administered to patients with bladder carcinoma and melanoma cancers. The most promising result was achieved in patients with stage 2 and 3 of ovarian carcinoma. The in vitro study of IFN-gamma in cancer cells is more extensive and results indicate anti-proliferative activity of IFN-gamma leading to the growth inhibition or cell death, generally induced by apoptosis but sometimes by autophagy.
Interleukin
Interleukins have an array of immune system effects. Interleukin-2 is used in the treatment of melanoma and renal cell carcinoma. In normal physiology it promotes both effector T cells and T-regulatory cells, but its exact mechanism of action is unknown.
Predictive genetic testing
Due to the high cost of immunotherapy and the reluctance of insurers to pre-authorize treatment, various genetic and molecular tests have been developed to predict therapeutic response. Three major biomarkers are currently FDA-approved and widely used in clinical practice: (1) programmed death-ligand 1 (PD-L1) expression, (2) microsatellite instability (MSI) or mismatch repair deficiency (dMMR), and (3) tumor mutational burden (TMB).
PD-L1 expression, detected via immunohistochemistry, may indicate which tumors are more likely to respond to immune checkpoint inhibitors by revealing the presence of proteins that help cancer cells evade immune surveillance. However, its predictive value is limited by variability in expression across tumor types, locations, and testing platforms. MSI and dMMR, identified through molecular or immunohistochemical testing, indicate a deficiency in DNA repair mechanisms and are associated with high mutation rates that may increase tumor immunogenicity. These biomarkers have been approved to guide the use of checkpoint inhibitors in several cancer types. TMB, measured by next-generation sequencing, quantifies the total number of somatic mutations in a tumor genome. High TMB has been associated with improved responses to immunotherapy, although its clinical utility remains controversial and context-dependent. As of 2023, reliance on TMB as a selection criterion for immunotherapy was still debated in the scientific community.
In addition to these biomarkers, the FDA has approved certain drug-companion diagnostic pairings based on genetic mutations. For example, BRAF-targeted therapies for metastatic melanoma are only indicated for patients whose tumors harbor a BRAF mutation.
While these tests are now commonly marketed as essential tools for precision oncology, they remain costly. Moreover, some have been the subject of controversy or misuse. Notable examples include the Duke University cancer fraud scandal and questionable claims surrounding so-called "liquid biopsies."
Research
Oncolytic virus
An oncolytic virus is a virus that preferentially infects and kills cancer cells. As the infected cancer cells are destroyed by oncolysis, they release new infectious virus particles or virions to help destroy the remaining tumour. Oncolytic viruses are thought not only to cause direct destruction of the tumour cells, but also to stimulate host anti-tumour immune responses for long-term immunotherapy.
The potential of viruses as anti-cancer agents was first realized in the early twentieth century, although coordinated research efforts did not begin until the 1960s. A number of viruses including adenovirus, reovirus, measles, herpes simplex, Newcastle disease virus and vaccinia have now been clinically tested as oncolytic agents. T-Vec is the first FDA-approved oncolytic virus for the treatment of melanoma. A number of other oncolytic viruses are in Phase II-III development.
Polysaccharides
Certain compounds found in mushrooms, primarily polysaccharides, can up-regulate the immune system and may have anti-cancer properties. For example, beta-glucans such as lentinan have been shown in laboratory studies to stimulate macrophage, NK cells, T cells and immune system cytokines and have been investigated in clinical trials as immunologic adjuvants.
Neoantigens
Main article: Neoantigen}}Many tumors express mutations. These mutations potentially create new targetable antigens (neoantigens) for use in T-cell immunotherapy. The presence of CD8+ T cells in cancer lesions, as identified using RNA sequencing data, is higher in tumors with a high [[tumor mutational burden
Polysaccharide-K
In the 1980s, Japan's Ministry of Health, Labour and Welfare approved polysaccharide-K extracted from the mushroom, Coriolus versicolor, to stimulate the immune systems of patients undergoing chemotherapy. It is a dietary supplement in the US and other jurisdictions.
Small molecule drugs
Small molecule drugs are also under development as cancer immunotherapeutic agents, offering potential advantages over traditional antibody-based therapies such as improved tissue penetration, oral bioavailability, and lower production costs. These molecules target key immune checkpoints and signaling pathways—including PD-1/PD-L1, STING, chemokine receptors, and negative regulators of T cell function—modulating the tumor microenvironment and enhancing anti-tumor immune responses. Notable candidates include BMS-202 and CA-170, which disrupt PD-1/PD-L1 interactions, and compounds targeting adenosine, prostaglandin, and innate immune pathways, many of which are advancing through clinical trials.
Cancer Resistance to Immunotherapy Treatment
While immunotherapy has provided a tool for combating cancer, a significant clinical challenge comes with it. Cancer cells can develop resistance to immunotherapy treatment, which decreases the efficacy of the treatment. A substantial proportion of patients either begin treatment with resistance, causing failure in response, or develop resistance after the medication is administered. A recent review estimated that a majority of patients with solid tumors eventually acquire resistance to immunotherapy.
Resistance to immunotherapy falls under either the category of primary or innate resistance, which causes a lack of response to immunotherapy treatment from the get-go, or acquired resistance, where there is an initial positive response to treatment followed by regression. Resistance arises through mechanisms that alter the interaction between tumor cells, the immune system, and the tumor microenvironment.
Tumor antigenicity, or the ability for the immune system to recognize cancer cells, is a primary target for resistance. Resistance mutations that down-regulate (decrease) the expression of antigen-presenting factors allow the immune system to ignore the presence of cancer cells. The major histocompatibility complex (MHC), which presents antigens on the cell surface to prompt immune recognition, is often a target. When T cells can no longer identify malignant cells, this renders therapies and immune recognition useless.
A second mechanism of immunotherapy resistance is impaired immune cell function. Cancers are found to attract and rely on immune suppressors such as regulatory T cells or other cells in the tumor microenvironment to decrease the response of immune cells. Further, reducing the activity of cytotoxic T cells by sustaining expression of inhibitory receptors limits the effectiveness of immune checkpoint inhibitors.
Cancers can modify the tumor microenvironment itself, creating physical and biochemical barriers to immune attack. Abnormal vasculature is a prominent way of doing this. Tumors secrete factors that attract the growth of blood vessels (angiogenesis) that encourages further methods of nutrient transport, feeding the tumor. Additionally, cancer cells can produce nerve growth factor which causes healthy neurons to develop around and within tumors. This is promotes further tumor growth because nerves produce neurotransmitters 5HT that prompt tumor cell proliferation. Essentially solid tumors are able to create networks of feedback loops, hijacking healthy cells.
References
References
- (14 December 2022). "Harnessing the immune system to develop breakthrough cancer therapies".
- (August 2021). "Targeting the Myeloid-Derived Suppressor Cell Compartment for Inducing Responsiveness to Immune Checkpoint Blockade Is Best Limited to Specific Subtypes of Gastric Cancers.". Gastroenterology.
- (April 2020). "A review of cancer immunotherapy: from the past, to the present, to the future". Current Oncology.
- (2022). "Clinical cancer immunotherapy: Current progress and prospects". Frontiers in Immunology.
- "Immunotherapy by Treatment Types".
- (August 2020). "The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications". Cellular & Molecular Immunology.
- "The Nobel Prize in Physiology or Medicine 2018".
- (April 2016). "Spontaneous regression of tumour and the role of microbial infection--possibilities for cancer treatment". Anti-Cancer Drugs.
- (March 2012). "Fever in Cancer Treatment: Coley's Therapy and Epidemiologic Observations". Global Advances in Health and Medicine.
- (2006). "The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas". The Iowa Orthopaedic Journal.
- "Commencement speakers praise, advise local graduates . . .". Washington Post.
- "Development of Cancer Immunotherapy". [[National Cancer Institute]].
- (24 September 2019). "Immunotherapy to Treat Cancer". [[National Cancer Institute]].
- (29 May 2024). "Immunotherapy for Cancer: An Overview".
- (December 2014). "Classification of current anticancer immunotherapies". Oncotarget.
- "Types of Biological Therapy". [[National Cancer Institute]].
- (30 September 2013). "What are Cancer Vaccines?".
- (July 2001). "Progress in cancer vaccines by enhanced self-presentation". Proceedings of the National Academy of Sciences of the United States of America.
- (July 2013). "Dendritic-cell-based therapeutic cancer vaccines". Immunity.
- (July 2016). "The present status and future prospects of peptide-based cancer vaccines". International Immunology.
- (June 2018). "eLS". American Cancer Society.
- (April 2012). "Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer". Human Vaccines & Immunotherapeutics.
- (May 2013). "Progress in emerging therapies for advanced prostate cancer". Cancer Treatment Reviews.
- (June 2012). "Development of sipuleucel-T: autologous cellular immunotherapy for the treatment of metastatic castrate-resistant prostate cancer". Vaccine.
- (January 2013). "Building on sipuleucel-T for immunologic treatment of castration-resistant prostate cancer". Cancer Control.
- (March 2012). "Adoptive immunotherapy for cancer: harnessing the T cell response". Nature Reviews. Immunology.
- (22 September 2021). "γδ T Cells for Leukemia Immunotherapy: New and Expanding Trends". Frontiers in Immunology.
- (February 2014). "Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells". Journal of Translational Medicine.
- Office of the Commissioner. "Press Announcements – FDA approval brings first gene therapy to the United States".
- (18 October 2017). "FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma". fda.gov.
- (29 March 2022). "Implantable immunotherapy "factory" fights cancer faster, more effectively".
- (March 2022). "Bioinstructive implantable scaffolds for rapid in vivo manufacture and release of CAR-T cells". Nature Biotechnology.
- (August 2015). "Targeted Activation of Toll-Like Receptors: Conjugation of a Toll-Like Receptor 7 Agonist to a Monoclonal Antibody Maintains Antigen Binding and Specificity". Bioconjugate Chemistry.
- (August 2014). "Type I and type II Fc receptors regulate innate and adaptive immunity". Nature Immunology.
- (June 2012). "Safety, activity, and immune correlates of anti-PD-1 antibody in cancer". The New England Journal of Medicine.
- (October 2015). "FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis". Cancer Cell.
- (May 2017). "In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy". Science Translational Medicine.
- (July 2016). "Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcγR Engagement". Cancer Cell.
- (May–Jun 2010). "The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions". mAbs.
- (May 2010). "Monoclonal antibodies: versatile platforms for cancer immunotherapy". Nature Reviews. Immunology.
- (2013). "Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies". Frontiers in Immunology.
- (June 2010). "Macrophages as mediators of tumor immunosurveillance". Trends in Immunology.
- (May 2017). "Cancer immunotherapy targeting the CD47/SIRPα axis". European Journal of Cancer.
- (March 2017). "The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer". Immunological Reviews.
- (March 2018). "SIRPα-CD47 Immune Checkpoint Blockade in Anticancer Therapy". Trends in Immunology.
- (January 2014). "Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy". FEBS Letters.
- (March 2004). "Complement function in mAb-mediated cancer immunotherapy". Trends in Immunology.
- (June 2024). "A spatial architecture-embedding HLA signature to predict clinical response to immunotherapy in renal cell carcinoma". Nature Medicine.
- (March 2012). "Antibody therapy of cancer". Nature Reviews. Cancer.
- (March 2003). "Immunotherapy: past, present and future". Nature Medicine.
- (February 2008). "FDA drug approval summary: alemtuzumab as single-agent treatment for B-cell chronic lymphocytic leukemia". The Oncologist.
- (18 May 2016). "FDA approves new, targeted treatment for bladder cancer". FDA.
- (12 September 2024). "FDA approves atezolizumab and hyaluronidase-tqjs".
- (12 September 2024). "FDA Approves Genentech's Tecentriq Hybreza, the First and Only Subcutaneous Anti-PD-(L)1 Cancer Immunotherapy".
- (12 September 2024). "Halozyme Announces FDA Approval of Roche's Tecentriq Hybreza With Enhanze for Multiple Types of Cancer". Halozyme Therapeutics.
- "US Food and Drug Administration – Avelumab Prescribing Label".
- Center for Drug Evaluation and Research. "Approved Drugs – Durvalumab (Imfinzi)".
- (9 February 2019). "FDA approves durvalumab after chemoradiation for unresectable stage III NSCLC". FDA.
- "Bristol-Myers Squibb and AbbVie Receive U.S. FDA Breakthrough Therapy Designation for Elotuzumab, an Investigational Humanized Monoclonal Antibody for Multiple Myeloma | BMS Newsroom".
- "FDA approval for Ipilimumab".
- (April 2015). "The future of immune checkpoint therapy". Science.
- "Opdivo (nivolumab) FDA Approval History".
- (September 2010). "U.S. Food and Drug Administration approval: ofatumumab for the treatment of patients with chronic lymphocytic leukemia refractory to fludarabine and alemtuzumab". Clinical Cancer Research.
- (20 December 2019). "FDA approves pembrolizumab in combination with chemotherapy for first-line treatment of metastatic squamous NSCLC". FDA.
- (9 February 2019). "Pembrolizumab (KEYTRUDA) for classical Hodgkin lymphoma". FDA.
- (20 December 2019). "FDA approves pembrolizumab for Merkel cell carcinoma". FDA.
- (9 February 2019). "FDA approves pembrolizumab for treatment of relapsed or refractory PMBCL". FDA.
- (18 September 2014). "National Cancer Institute – Pembrolizumab Use in Cancer".
- (December 1997). "FDA approves new kind of lymphoma treatment. Food and Drug Administration". AIDS Treatment News.
- (8 July 2024). "Rituxan Hycela – rituximab and hyaluronidase injection, solution".
- (1 January 2004). "Chronic lymphocytic leukemia". Hematology. American Society of Hematology. Education Program.
- (2001). "CD52 antigen--a review". Medical Science Monitor.
- (July 2012). "How I treat prolymphocytic leukemia". Blood.
- (9 February 2019). "FDA approves durvalumab after chemoradiation for unresectable stage III NSCLC". FDA.
- (June 2011). "Ipilimumab". Nature Reviews. Drug Discovery.
- (November 2011). "Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma". Clinical Cancer Research.
- (December 2010). "Ipilimumab: a promising immunotherapy for melanoma". Oncology.
- (2001). "CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy". Annual Review of Immunology.
- (2017). "Current Diagnosis and Management of Immune Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitor Therapy". Frontiers in Pharmacology.
- (2010). "The role of ofatumumab in the treatment of chronic lymphocytic leukemia resistant to previous therapies". Journal of Blood Medicine.
- (Jul–Aug 2009). "Ofatumumab". mAbs.
- (May 2017). "Pembrolizumab label". FDA.
- (27 January 2017). "Pembrolizumab label at eMC". UK Electronic Medicines Compendium.
- (June 2018). "HIGHLIGHTS OF PRESCRIBING INFORMATION – KEYTRUDA (Pembrolizumab)".
- (July 2010). "Rituximab: a review of its use in chronic lymphocytic leukaemia, low-grade or follicular lymphoma and diffuse large B-cell lymphoma". Drugs.
- (2003). "Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia". Drugs.
- (November 2002). "Mechanism of action of rituximab". Anti-Cancer Drugs.
- (February 2018). "Immunobiology". Garland Science.
- (April 2010). "Rituximab: mechanism of action". Seminars in Hematology.
- (March 2012). "The blockade of immune checkpoints in cancer immunotherapy". Nature Reviews. Cancer.
- (9 March 2020). "Biomarkers for Response to Immune Checkpoint Blockade". Annual Review of Cancer Biology.
- (October 2023). "Impact of Helicobacter pylori infection status on outcomes among patients with advanced gastric cancer treated with immune checkpoint inhibitors.". J Immunother Cancer.
- (January 2024). "Impact of Helicobacter pylori infection status on outcomes among patients with advanced gastric cancer treated with immune checkpoint inhibitors.". J Immunother Cancer.
- (2017). "Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer". ESMO Open.
- (May 2011). "Ipilimumab: first global approval". Drugs.
- (June 2024). "Current and future immunotherapeutic approaches in pancreatic cancer treatment". Journal of Hematology & Oncology.
- (June 2012). "Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study". Journal of Clinical Oncology.
- (September 2013). "Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer". Journal of Immunotherapy.
- {{ClinicalTrialsGov. NCT01928394. A Study of Nivolumab by Itself or Nivolumab Combined With Ipilimumab in Patients With Advanced or Metastatic Solid Tumors
- (June 2015). "Immune Checkpoint Blockade in Cancer Therapy". Journal of Clinical Oncology.
- (April 2024). "Immune Checkpoint Inhibitor Use During Pregnancy and Outcomes in Pregnant Individuals and Newborns". JAMA Network Open.
- (February 2017). "Local checkpoint inhibition of CTLA-4 as a monotherapy or in combination with anti-PD1 prevents the growth of murine bladder cancer". European Journal of Immunology.
- (18 May 2016). "F.D.A. Approves an Immunotherapy Drug for Bladder Cancer". The New York Times.
- (5 August 2016). "Bristol Myers: Opdivo Failed to Meet Endpoint in Key Lung-Cancer Study". The Wall Street Journal.
- BeiGene, Ltd.. (2016). "BeiGene Presents Initial Clinical Data on PD-1 Antibody BGB-A317 at the 2016 American Society of Clinical Oncology Annual Meeting". Globe Newswire.
- "FDA grants priority review for Roche's cancer immunotherapy atezolizumab in specific type of lung cancer". Roche.
- "Immuno-oncology Avelumab". Merck Group.
- (April 2016). "Durvalumab continues to progress in treatment of advanced bladder cancer.". Cure today.
- "Affimer biotherapeutics target cancer's off-switch with PD-L1 inhibitor". Avacta Life Sciences.
- (February 2016). "Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy". Immunity.
- (2017). "Combination immunotherapy: a road map". Journal for Immunotherapy of Cancer.
- (August 2015). "Combination cancer immunotherapy and new immunomodulatory targets". Nature Reviews. Drug Discovery.
- (2015). "Thermal Ablative Therapies and Immune Checkpoint Modulation: Can Locoregional Approaches Effect a Systemic Response?". Gastroenterology Research and Practice.
- (January 2018). "Comprehensive analysis of the clinical immuno-oncology landscape". Annals of Oncology.
- (March 2018). "Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity". The Journal of Experimental Medicine.
- (21 May 2018). "TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy". Nature Biomedical Engineering.
- (June 2024). "JAK inhibition enhances checkpoint blockade immunotherapy in patients with Hodgkin lymphoma". Science.
- (January 2004). "Cytokines in cancer pathogenesis and cancer therapy". Nature Reviews. Cancer.
- "Immunotherapy For Cancer".
- (November 2006). "Interferons, immunity and cancer immunoediting". Nature Reviews. Immunology.
- (2011). "Interferon lambda: a new sword in cancer immunotherapy". Clinical & Developmental Immunology.
- (December 2016). "Review of the recombinant human interferon gamma as an immunotherapeutic: Impacts of production platforms and glycosylation". Journal of Biotechnology.
- (2012). "The 20th anniversary of interleukin-2 therapy: bimodal role explaining longstanding random induction of complete clinical responses". Cancer Management and Research.
- (2021). "FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients". Frontiers in Oncology.
- "Cancer Immunotherapy Biomarker Testing: What Pathologists Need to Know".
- (3 February 2016). "Cancer Genetics offers the FDA-approved DAKO PD-L1 IHC 22C3 pharmDx companion diagnostic test for KEYTRUDA®". Cancer Genetics Inc..
- (February 2018). "PD-L1 diagnostic tests: a systematic literature review of scoring algorithms and test-validation metrics". Diagnostic Pathology.
- (April 2018). "Time is up for PD-L1 testing standardization in lung cancer". Annals of Oncology.
- (November 2017). "Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers". Molecular Cancer Therapeutics.
- (7 February 2018). "FDA Accepts sBLA for First-Line Nivolumab Plus Low-Dose Ipilimumab in NSCLC With Tumor Mutational Burden ≥ 10 mut/mb". [[American Society of Clinical Oncology]].
- (February 2025). "Tumor mutational burden: why is it still a controversial agnostic immunotherapy biomarker?". Future Oncology.
- (October 2024). "Is tumor mutational burden predictive of response to immunotherapy?". eLife.
- (27 June 2018). "FDA approves Encorafenib and Binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations". U.S. Food and Drug Administration.
- (April 2024). "Cost-Effectiveness and the Economics of Genomic Testing and Molecularly Matched Therapies". Surgical Oncology Clinics of North America.
- (22 January 2015). "Duke U Cancer Fraud Scandal: A Cautionary Tale For Obama's Precision Medicine Push". Forbes.
- [https://sciencebasedmedicine.org/liquid-biopsies-for-cancer-life-saving-tests-or-overdiagnosis-and-overtreatment-taken-to-a-new-level/ "Liquid biopsies" for cancer screening: Life-saving tests, or overdiagnosis and overtreatment taken to a new level?] David Gorski, 28 September 2015, [[Science-Based Medicine]] website
- [https://www.melanoma.org/find-support/patient-community/mpip-melanoma-patients-information-page/insurance-wont-pay-braf-test A public discussion by cancer patients] {{Webarchive. link. (3 July 2015 from 2011 on the melanoma.org website shows costs and claims.)
- (October 2016). "Oncolytic virus therapy: A new era of cancer treatment at dawn". Cancer Science.
- (2017). "Genetically Engineered Vaccinia Viruses As Agents for Cancer Treatment, Imaging, and Transgene Delivery". Frontiers in Oncology.
- (January 2018). "Cancer immunotherapy beyond immune checkpoint inhibitors". Journal of Hematology & Oncology.
- (June 2017). "Oncolytic Viruses in Cancer Treatment: A Review". JAMA Oncology.
- (June 2013). "β-Glucans and their applications in cancer therapy: focus on human studies". Anti-Cancer Agents in Medicinal Chemistry.
- (December 2014). "Genetic basis for clinical response to CTLA-4 blockade in melanoma". The New England Journal of Medicine.
- (April 2015). "Neoantigens in cancer immunotherapy". Science.
- "Coriolus Versicolor". American Cancer Society.
- (September 2024). "Small-molecule in cancer immunotherapy: Revolutionizing cancer treatment with transformative, game-changing breakthroughs". Biochimica et Biophysica Acta (BBA) - Reviews on Cancer.
- (2023). "Small molecule inhibitors for cancer immunotherapy and associated biomarkers – the current status". Frontiers in Immunology.
- (January 2019). "The Next Generation of Immunotherapy for Cancer: Small Molecules Could Make Big Waves". Journal of Immunology.
- Said SS, Ibrahim WN. "Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives". ''Pharmaceutics''. 2023;15(4):1143. doi:10.3390/pharmaceutics15041143.
- Verdys P, Johansen AZ, Gupta A, Presti M, Dionisio E, Madsen DH, Curioni-Fontecedro A, Donia M. "Acquired resistance to immunotherapy in solid tumors". ''Trends in Molecular Medicine''. 2025.
- Alsaafeen BH, Ali BR, Elkord E. "Resistance mechanisms to immune checkpoint inhibitors: Updated insights". ''Molecular Cancer''. 2025;24:20. doi:10.1186/s12943-024-02212-7.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about Cancer immunotherapy — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report