From Surf Wiki (app.surf) — the open knowledge base
Beta thalassemia
Hereditary blood disorder causing anemia
Hereditary blood disorder causing anemia
| Field | Value |
|---|---|
| name | Beta-thalassemia |
| synonyms | Cooley's anemia, Mediterranean anemia |
| image | Thalassemia beta.jpg |
| caption | Beta-thalassemia genetics, the picture shows one example of how beta-thalassemia is inherited. The beta-globin gene is located on chromosome 11. A child inherits two beta globin genes (one from each parent). |
| field | Hematology |
| symptoms | Anemia, enlarged spleen, abnormal bone structure |
| onset | Between 6 and 24 months of age |
| types | Beta-thalassemia minor, intermedia and major |
| causes | Mutations in the HBB gene |
| risks | Family history, ancestry from the Mediterranean, West Africa, the Middle East, the Indian subcontinent or Southeast Asia |
| diagnosis | Blood smear, hemoglobin electrophoresis, iron & ferritin tests, DNA analysis |
| differential | Alpha thalassemia, sickle cell disease, iron deficiency anemia |
| prevention | Preconception genetic counseling |
| treatment | Blood transfusion, iron chelation, stem cell transplant, gene therapy |
Beta-thalassemia (β-thalassemia) is an inherited blood disorder, a form of thalassemia resulting in variable outcomes ranging from clinically asymptomatic to severe anemia individuals. It is caused by reduced or absent synthesis of the beta chains of hemoglobin, the molecule that carries oxygen in the blood. Symptoms depend on the extent to which hemoglobin is deficient, and include anemia, pallor, tiredness, enlargement of the spleen, jaundice, and gallstones. In severe cases death ensues.
Beta thalassemia occurs due to a mutation of the HBB gene leading to deficient production of the hemoglobin subunit beta-globin; the severity of the disease depends on the nature of the mutation, and whether or not the mutation is homozygous. The body's inability to construct beta-globin leads to reduced or zero production of adult hemoglobin thus causing anemia. The other component of hemoglobin, alpha-globin, accumulates in excess leading to ineffective production of red blood cells, increased hemolysis, and iron overload. Diagnosis is by checking the medical history of near relatives, microscopic examination of blood smear, ferritin test, hemoglobin electrophoresis, and DNA sequencing.
As an inherited condition, beta thalassemia cannot be prevented although genetic counselling of potential parents prior to conception can propose the use of donor sperm or eggs. Patients may require repeated blood transfusions throughout life to maintain sufficient hemoglobin levels; this in turn may lead to severe problems associated with iron overload. Medication includes folate supplementation, iron chelation, bisphosphonates, and removal of the spleen. Beta thalassemia can also be treated by bone marrow transplant from a well matched donor, or by gene therapy.
Thalassemias were first identified in severely sick children in 1925, with identification of alpha and beta subtypes in 1965. Beta-thalassemia tends to be most common in populations originating from the Mediterranean, the Middle East, Central and Southeast Asia, the Indian subcontinent, and parts of Africa. This coincides with the historic distribution of Plasmodium falciparum malaria, and it is likely that a hereditary carrier of a gene for beta-thalassemia has some protection from severe malaria. However, because of population migration, β-thalassemia can be found around the world. In 2005, it was estimated that 1.5% of the world's population are carriers and 60,000 affected infants are born with the thalassemia major annually.
Signs and symptoms


Symptoms depend on the type and severity of thalassemia. Carriers of thalassemia genes may have no symptoms (thalassemia minor) or very mild symptoms with occasional crisis (thalassemia intermedia); individuals who are homozygous for the mutation have severe and life threatening symptoms (thalassemia major).
Individuals with beta-thalassemia major usually present within the first two years of life with symptomatic severe anemia, poor growth, and skeletal abnormalities. Untreated thalassemia major eventually leads to death, usually by heart failure.
Those with beta-thalassemia intermedia (those who are compound heterozygotes for the beta thalassemia mutation) usually present later in life with mild to moderate symptoms of anemia.
Beta thalassemia trait (beta thalassemia minor) involves heterozygous inheritance of a beta-thalassemia mutation. Individuals usually have microcytosis with mild anemia; they are usually asymptomatic or have mild symptoms. Beta thalassemia minor can also present as beta-thalassemia silent carriers; those who inherit a beta thalassemic mutation but have no hematologic abnormalities or symptoms.
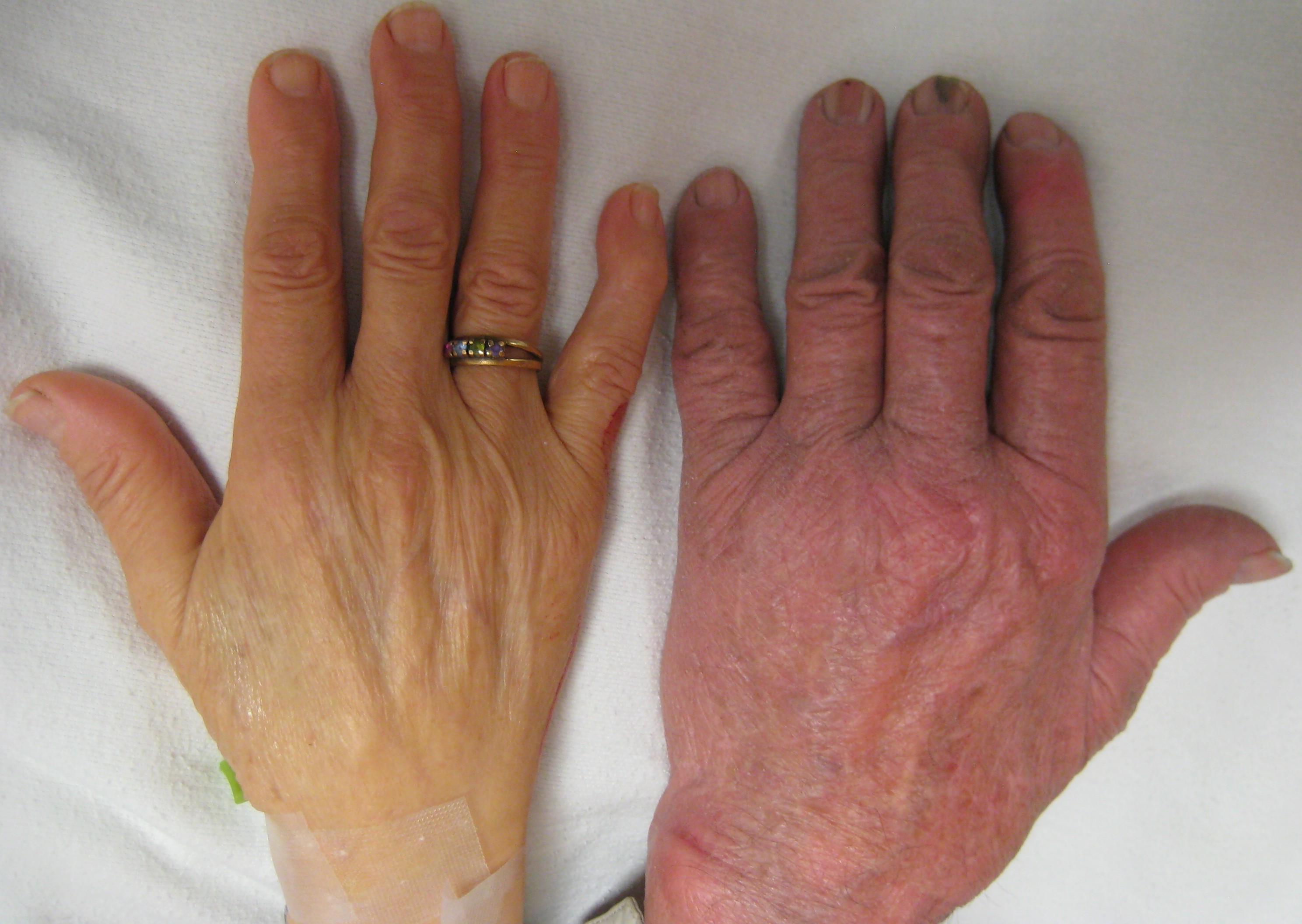
Individuals with thalassemia thalassemia major and intermedia (to a lesser extent) are susceptible to health complications that involve the spleen (hypersplenism) and gallstones (due to hyperbilirubinemia from peripheral hemolysis). Additional symptoms of beta-thalassemia major or intermedia include the classic symptoms of anemia including fatigue, developmental delay in childhood, leg ulcers, and organ failure. Ineffective erythropoiesis (red blood cell production) can lead to expansion of the bone marrow in compensation; this can then lead to deformity, bone pain, and craniofacial abnormalities. Organs such as the liver and spleen that can also become enrolled in red blood cell production, leading to hepatosplenomegaly (enlargement of the liver and spleen).
People with thalassemia can get too much iron in their bodies, either from the disease itself as RBCs are destroyed, or as a consequence of frequent blood transfusions. Excess iron is not excreted, but forms toxic non-transferrin-bound iron. This can lead to organ damage, potentially affecting the heart, liver, endocrine system, bones and spleen. Symptoms include an irregular heartbeat, cardiomyopathy, cirrhosis of the liver, hypothyroidism, delayed puberty and fertility problems, brittle and deformed bones, and an enlarged spleen.
For clinical purposes, thalassemia is categorised as either transfusion-dependent thalassemia (TDT) or non-transfusion-dependent thalassemia (NTDT) are used. Patients are usually considered as having NTDT if they have received fewer than 6 red blood cell units in the past 6 months and none in the preceding 2 months.
Cause

Hemoglobin structural biology
Mutations
β-globin chains are encoded by the HBB gene on chromosome 11; in a healthy person with two copies on each chromosome, two loci encode the β chain. In beta thalassemia, a single faulty gene can be either asymptomatic or cause mild disease; if both genes are faulty this causes moderate to severe disease.
More than 350 mutations have been identified which can cause beta thalassemia; 20 of these account for 80% of beta-thalassemia cases.
Two major groups of mutations can be distinguished:
- Nondeletion forms: These defects, in general, involve a single base-pair substitution or small insertions near or upstream of the HBB gene.
- Deletion forms: base-pair deletions of different sizes involving the HBB gene produce syndromes such as hereditary persistence of fetal hemoglobin syndrome. Mutations are characterized as (βo) if they prevent any formation of β globin chains, and mutations are characterized as (β+) if they allow some β globin chain formation to occur.
| Name | Older synonyms | Description | Alleles |
|---|---|---|---|
| Thalassemia minor | Heterozygous form: Only one of the β globin alleles bears a mutation. Affected individuals will develop microcytic anemia. Detection usually involves a lower than normal mean corpuscular volume value ( | β+/β | |
| βo/β | |||
| Thalassemia intermedia | Affected individuals can often manage a normal life but may need occasional transfusions, e.g., at times of illness or pregnancy, depending on the severity of their anemia. | β+/β+ | |
| βo/β+ | |||
| Thalassemia major | Mediterranean anemia; Cooley anemia | Homozygous form: Occurs when both alleles have thalassemia mutations. Untreated, it causes severe anemia, splenomegaly and bone deformities, and progresses to death before age 20. Treatment consists of periodic blood transfusion; splenectomy for splenomegaly and chelation of transfusion-related iron overload. | βo/βo |
Due to globin defects, beta thalassemia patients do not have normal levels of adult hemoglobin (HbA), and instead have elevated levels of HbA2 (α2δ2). Production of this form of hemoglobin may increase as a consequence of stress erythropoiesis.
Diagnosis
Prevention
Risk factors
Family history and ancestry are factors that increase the risk of beta-thalassemia. Depending on family history, if a person's parents or grandparents had beta thalassemia major or intermedia, there is a 75% (3 out of 4) probability (see inheritance chart at top of page) of the mutated gene being inherited by an offspring. Even if a child does not have symptomatic beta thalassemia they can still be a carrier, leading to an increased risk in future generations of their offspring having beta-thalassemia.
Beta thalassemia occurs most often in people of Mediterranean, Middle Eastern, Southern Asian, and African ancestry.
Counselling and screening
The American College of Obstetricians and Gynecologists recommends all people thinking of becoming pregnant should be offered testing to see if they have thalassemia trait. Genetic counseling and genetic testing are recommended for families who carry a thalassemia trait. Understanding the genetic risk, ideally before a family is started, would hopefully allow families to understand more about the condition and make an informed decision that is best for their family.
A number of countries have programs aimed at reducing the incidence of beta-thalassemia:-
- Cyprus has one of the highest carrier rates in the world. A program of premarital screening and counselling has, since the program's implementation in the 1970s, reduced the number of children born with thalassemia major from one of every 158 births to almost zero. Greece also has a screening program to identify people who are carriers.
- In Iran as a premarital screening, the man's red cell indices are checked first. If he has microcytosis (mean cell hemoglobin
- Large-scale awareness campaigns are being organized in India both by government and non-government organizations to promote voluntary premarital screening, with marriage between carriers strongly discouraged.
Treatment
Main article: Management of thalassemia}}{{#section:Thalassemia, Thal_Management
Combination hemoglobinopathies
A combination hemoglobinopathy occurs when someone inherits two different abnormal hemoglobin genes. If these are different versions of the same gene, one having been inherited from each parent it is an example of compound heterozygosity.
Some examples of clinically significant combinations involving beta thalassemia include:
-
Hemoglobin C/ beta thalassemia: common in Mediterranean and African populations generally results in a moderate form of anemia with splenomegaly.
-
Hemoglobin D/ beta thalassemia: common in the northwestern parts of India and Pakistan (Punjab region).
-
Hemoglobin E/ beta thalassemia: common in Cambodia, Thailand, and parts of India, it is clinically similar to β thalassemia major or β thalassemia intermedia.
-
Hemoglobin S/ beta thalassemia: common in African and Mediterranean populations, it is clinically similar to sickle-cell anemia.
-
Delta-beta thalassemia is a rare form of thalassemia in which there is a reduced production of both the delta and beta globins. It is generally asymptomatic.
Epidemiology
Beta thalassemia is particularly prevalent among the Mediterranean peoples and this geographical association is responsible for its naming: thalassa (θάλασσα) is the Greek word for sea and haima (αἷμα) is the Greek word for blood. In Europe, the highest prevalence of beta-thalassemia trait is found in Greece, Turkey, and Mediterranean islands such as Sicily, Sardinia, Corsica, Cyprus, Malta and Crete.
Incidence
Beta thalassemia is most prevalent in the "thalassemia belt" which includes areas in Sub-Saharan Africa, and the Mediterranean extending into the Middle East and Southeast Asia. This geographical distribution is thought to be due to the beta-thalassemia carrier state (beta-thalassemia minor) conferring resistance to malaria. In 2005, it was estimated that 1.5% of the world's population are carriers and 60,000 affected infants are born with the thalassemia major annually.
Evolutionary adaptation
The thalassemia trait may confer a degree of protection against malaria, which is historically endemic in the regions where the trait is common. This is thought to confer a selective survival advantage on carriers (known as heterozygous advantage), thus perpetuating the mutation. In that respect, the various thalassemias resemble other genetic disorders affecting hemoglobin, such as sickle-cell disease or Hemoglobin C disease.
References
References
- "Beta Thalassemia Treatment & Management".
- (10 November 2009). "Evolution of hemoglobinopathy prevention in Africa: results, problems and prospect". Mediterranean Journal of Hematology and Infectious Diseases.
- (December 2020). "Changing patterns in the epidemiology of β-thalassemia". European Journal of Haematology.
- (2014-04-17). "Maternal-Child Nursing". Elsevier Health Sciences.
- "Lanzkowsky's Manual Of Pediatric Hematology And Oncology 6th Edition ( 2016)".
- "Beta Thalassemia".
- (2015-04-21). "Goldman-Cecil Medicine: Expert Consult — Online". Elsevier Health Sciences.
- (2025). "StatPearls". StatPearls Publishing.
- (2012-02-16). "Oxford Handbook of Clinical Pathology". OUP Oxford.
- (June 2022). "Thalassaemia". Lancet.
- (8 February 2024). "GeneReviews®". University of Washington, Seattle.
- (17 October 2022). "Thalassaemia - Thalassaemia carriers".
- (May 2010). "Beta-thalassemia". Orphanet Journal of Rare Diseases.
- (27 January 2025). "Alpha Thalassemia".
- (2023-11-10). "Clinical commissioning policy: allogeneic haematopoietic stem cell transplantation (Allo-HSCT) for adult transfusion dependent thalassaemia".
- (16 November 2023). "MHRA authorises world-first gene therapy that aims to cure sickle-cell disease and transfusion-dependent β-thalassemia".
- Stuart H. Orkin. (2009). "Nathan and Oski's Hematology of Infancy and Childhood, Volume 1". Elsevier Health Sciences.
- (May 2010). "Alpha-thalassaemia". Orphanet Journal of Rare Diseases.
- (17 Nov 2021). "Thalassemia-Thalassemia - Symptoms & causes".
- (February 2021). "β-Thalassemias". The New England Journal of Medicine.
- (2011). "Introduction to Pathology for the Physical Therapist Assistant". Jones & Bartlett Publishers.
- "Beta thalassemia".
- (June 2022). "Non-transferrin bound iron". Clinica Chimica Acta; International Journal of Clinical Chemistry.
- (January 2016). "Endocrine Problems in Thalassemia".
- (25 Oct 2023). "Thalassaemia".
- (September 2023). "How I treat non-transfusion-dependent β-thalassemia". Blood.
- {{OMIM. 141900. Hemoglobin—Beta Locus; HBB
- Robbins Basic Pathology, Page No:428
- (May 2013). "The molecular basis of β-thalassemia". Cold Spring Harbor Perspectives in Medicine.
- (February 2024). "Global distribution of β-thalassemia mutations: An update". Gene.
- (2007-11-25). "Molecular Pathology in Clinical Practice". Springer.
- (2012-12-06). "Contemporary Internal Medicine: Clinical Case Studies". Springer.
- National Organization for Rare Disorders. (2003). "NORD Guide to Rare Disorders". Lippincott Williams & Wilkins.
- (2000-01-13). "Hemochromatosis: Genetics, Pathophysiology, Diagnosis and Treatment". Cambridge University Press.
- ((Lippincott Williams & Wilkins)). (2009). "Professional Guide to Diseases". Lippincott Williams & Wilkins.
- (2020-09-01). "Stress erythropoiesis: definitions and models for its study". Experimental Hematology.
- "Risk Factors".
- (October 2022). "Carrier Screening for Hemoglobinopathies: Sickle Cell Disease and Thalassemia".
- (October 2021). "Preconception risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay-Sachs disease". The Cochrane Database of Systematic Reviews.
- (April 2005). "Thalassaemia screening in pregnancy". Current Opinion in Obstetrics & Gynecology.
- (October 2010). "Carrier screening for beta-thalassaemia: a review of international practice". European Journal of Human Genetics.
- (October 2011). "Haemoglobinopathies in Greece: prevention programme over the past 35 years". The Indian Journal of Medical Research.
- (November 2004). "Iranian national thalassaemia screening programme". BMJ.
- (January 2010). "Screening for beta thalassaemia". Indian Journal of Human Genetics.
- (December 2021). "Shared molecular basis, diagnosis, and co-inheritance of alpha and beta thalassemia". Blood Research.
- (April 2006). "Co-inheritance of alpha+-thalassaemia and sickle trait results in specific effects on haematological parameters". British Journal of Haematology.
- (February 2011). "Hemoglobin C".
- (March 2015). "Hemoglobin D-Punjab: origin, distribution and laboratory diagnosis". Revista Brasileira de Hematologia e Hemoterapia.
- (May 2008). "Studies in haemoglobin E beta-thalassaemia". British Journal of Haematology.
- (April 2024). "Hemoglobin S–Beta-Thalassemia Disease - Hematology and Oncology".
- (2005). "Textbook Of Practical Physiology". Orient Blackswan.
- {{OEtymD. thalassemia
- {{LSJ. qa/lassa. θάλασσα, {{LSJ. αἷμα. ref.
- "WHO {{!}} Global epidemiology of haemoglobin disorders and derived service indicators".
- (2013-10-16). "The MGH Review of Critical Care Medicine". Lippincott Williams & Wilkins.
- (2013). "Basic Genetics: A Primer Covering Molecular Composition of Genetic Material, Gene Expression and Genetic Engineering, and Mutations and Human Genetic". Universal-Publishers.
- (2010). "Williams Hematology". The McGraw-Hill Companies.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about Beta thalassemia — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report