From Surf Wiki (app.surf) — the open knowledge base
Alpha-thalassemia
Inherited blood disorder causing anemia

Inherited blood disorder causing anemia
| Field | Value |
|---|---|
| name | Alpha-thalassemia |
| synonyms | α-thalassemia |
| image | File:Alpha Thalassemia.jpg |
| caption | A microscope image of a giemsa-stained blood film from a person with alpha thalassemia (haemoglobin H (HbH) disease) |
| symptoms | Anemia, jaundice, enlarged spleen |
| complications | Iron overload |
| onset | Inherited - present in utero |
| causes | Genetically determined deficiency in alpha globin production |
| diagnosis | Blood smear, hemoglobin electrophoresis, DNA sequencing |
| differential | Beta thalassemia, iron deficiency anemia |
| treatment | Blood transfusion, possible splenectomy, bone marrow transplant |
Alpha-thalassemia (α-thalassemia, α-thalassaemia) is an inherited blood disorder and a form of thalassemia. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Symptoms depend on the extent to which hemoglobin is deficient, and include anemia, pallor, tiredness, enlargement of the spleen, iron overload, abnormal bone structure, jaundice, and gallstones. In severe cases death ensues, often in infancy, or death of the unborn fetus.
The disease is characterised by reduced production of the alpha-globin component of hemoglobin, caused by inherited mutations affecting the genes HBA1 and HBA2. This causes reduced levels of hemoglobin leading to anemia, while the accumulation of surplus beta-globin, the other structural component of hemoglobin, damages red blood cells and shortens their life. Diagnosis is by checking the medical history of near relatives, microscopic examination of blood smear, ferritin test, hemoglobin electrophoresis, and DNA sequencing.
As an inherited condition, alpha thalassemia cannot be prevented although genetic counselling of parents prior to conception can propose the use of donor sperm or eggs. The principle form of management is blood transfusion every 3 to 4 weeks, which relieves the anemia but leads to iron overload and possible immune reaction. Medication includes folate supplementation, iron chelation, bisphosphonates, and removal of the spleen. Alpha thalassemia can also be treated by bone marrow transplant from a well matched donor. Thalassemias were first identified in severely sick children in 1925, with identification of alpha and beta subtypes in 1965. Alpha thalassemia has its greatest prevalence in populations originating from Southeast Asia, Mediterranean countries, Africa, the Middle East, India, and Central Asia. Having a mild form of alpha thalassemia has been demonstrated to protect against malaria and thus can be an advantage in malaria endemic areas.
Cause
Alpha-thalassemia is almost always inherited. It is a recessive trait - a single defective gene is insufficient to cause illness. Due to the involvement of four alpha globin genes, the inheritance pattern is complex, with varying severity depending on the number of gene mutations inherited from each parent.
Normal individuals carry 4 alpha-globin genes, comprising autosomal pairs of the HBA1 and HBA2 genes. There are approximately 130 known mutations which can cause alpha thalassemia, mainly comprising deletion of part or all of a gene which then fails to produce alpha globin. If either one gene or two out of the four is faulty, the remaining genes produce sufficient alpha globin for normal life. If three genes are faulty, the sole functioning gene produces relatively small quantities of alpha globin, causing anemia and HbH disease. Four faulty genes (and therefore zero alpha globin) is incompatible with life.
In rare cases alpha thalassemia can be acquired as a consequence of myelodysplastic cancer.
Pathophysiology
Symptoms


Symptoms depend on the type and severity of thalassemia. Carriers of thalassemia genes may have no symptoms or very mild symptoms with occasional crisis; those with three or more (out of four) affected genes will have severe and life threatening symptoms.
Full alpha thalassemia with all four genes failing to synthesise alpha-globin, is generally fatal to the unborn child. The absence of alpha globin means that zero functional hemoglobin is produced during gestation. Unmatched gamma globin chains cluster to form hemoglobin Bart's, which is ineffective at transporting oxygen. In this situation, a fetus will develop hydrops fetalis, a form of edema, which can be detected on prenatal ultrasound. The child will normally die before or shortly after birth, unless intrauterine blood transfusion is performed. Less severe alpha thalassemia may affect growth and development.
If thalassemia is untreated or undetected in the infant, this can lead to developmental issues such as slowed growth, delayed puberty, bone abnormalities, and intellectual impairment.
More generally, impaired production of hemoglobin causes anemia, resulting in tiredness and a general lack of energy, shortness of breath, rapid or irregular heartbeat, dizziness, pale skin, yellowing of the skin and eyes (jaundice).
In thalassemia, ineffective erythropoiesis causes the bone marrow to expand. This expansion is a compensatory response to the damage caused to red blood cells by the imbalanced production of globin chains. Bone marrow expansion can lead to abnormal bone structure, particularly in the skull and face. Expansion of the bone marrow in the developing child leads to a distinctive facial shape often referred to as "Chipmunk facies". Other skeletal changes include osteoporosis, growth retardation, and malformation of the spine.
People with thalassemia can get too much iron in their bodies, either from the disease itself as RBCs are destroyed, or as a consequence of frequent blood transfusions. Excess iron is not excreted, but forms toxic non-transferrin-bound iron. This can lead to organ damage, potentially affecting the heart, liver, endocrine system, bones and spleen. Symptoms include an irregular heartbeat, cardiomyopathy, cirrhosis of the liver, hypothyroidism, delayed puberty and fertility problems, brittle and deformed bones, and an enlarged spleen.
The spleen is the organ which removes damaged red blood cells from circulation; in thalassemia patients it is abnormally active, causing it to enlarge and possibly become hyperactive, a condition called hypersplenism.
The immune system can become compromised in a number of ways; anemia, iron overload, and hypersplenism may affect the immune response and increase the risk of severe infection.
Diagnosis
Prognosis
The prognosis for alpha thalassemia depends on the degree to which alpha globin production is affected. Those with mild alpha thalassemia, involving deletion of one or two alpha-globin genes, do not generally require treatment and have a normal life expectancy. Hemoglobin H disease, with three of the four genes either deleted or inactive, gives a mild to moderate form of anemia but may lead normal lives.
The prognosis when all four genes are affected, leading to Hb Bart's hydrops fetalis, is very poor, with most affected fetuses dying in utero or shortly after birth due to severe fetal hypoxia. It can be treated with intrauterine transfusions, however survival remains low and the infant requires lifelong blood transfusions. As of 2017, 69 patients were known who have survived past infancy.
Treatment
Main article: Management of thalassemia
Treatment for thalassemia depends on the severity of the disease. People with thalassemia traits (thalassemia minor or non transfusion dependent thalassemia), may not require medical or follow-up care after the initial diagnosis is made. Occasionally transfusions may be necessary particularly around childbirth, surgery, or if other conditions provoke anemia. A folic acid supplement may also be recommended.
For those with severe forms of thalassemia (thalassemia major, or transfusion-dependent thalassemia), the three principal treatments are red blood cell transfusions to relieve anemia, iron chelation to mitigate the side effects of transfusion, and folic acid supplementation to encourage the growth of new blood cells. Other forms of treatment available depending on individual circumstances.
Red blood cell transfusions
Blood transfusions are the main treatment approach for prolonging life. Donated healthy red blood cells have a functional life of 4 to 6 weeks before they wear out and are broken down in the spleen. Regular transfusions every three to four weeks are necessary in order to maintain hemoglobin at a healthy level. Transfusions come with risks including iron overload, the risk of acquiring infections, and the risk of immune reaction to the donated cells (alloimmunization).
Iron chelation
Multiple blood transfusions lead to severe iron overload, as the body eventually breaks down the hemoglobin in donated cells. This releases iron which it is unable to excrete. Iron overload may be treated by chelation therapy with the medications deferoxamine, deferiprone, or deferasirox. Deferoxamine is only effective as a daily injection, complicating its long-term use. Adverse effects include primary skin reactions around the injection site and hearing loss. Deferasirox and deferiprone are both oral medications, whose common side effects include nausea, vomiting and diarrhea.
Folic acid
Folate is a B group vitamin which is involved in the manufacture of red blood cells. Folate supplementation, in the form of folic acid, is often recommended in thalassemia.
Osteoporosis
People with thalassemia are at a higher risk of osteoporosis. Treatment options include bisphosphonates and zinc supplementation.
Removal of the spleen
The spleen is the organ which removes damaged or misshapen red blood cells from the circulation. In thalassemia, this can lead to the spleen becoming enlarged, a condition known as splenomegaly. Slight enlargement of the spleen is not a problem, however if it becomes extreme then surgical removal of the spleen (splenectomy) may be recommended.
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation (HSCT) is a potentially curative treatment for both alpha and beta thalassemia. It involves replacing the dysfunctional stem cells in the bone marrow with healthy cells from a well-matched donor. Cells are ideally sourced from human leukocyte antigen matched relatives; the procedure is more likely to succeed in children rather than adults.
The first HSC transplant for thalassemia was carried out in 1981 on a patient with beta thalassemia major. Since then, a number of patients have received bone marrow transplants from healthy matched donors, although this procedure has a high level of risk.
In 2018 an unborn child with hydrops fetalis, a potentially fatal complication of alpha thalassemia, was successfully transfused in utero with her mother's stem cells.
HSCT is a dangerous procedure with many possible complications; it is reserved for patients with life-threatening diseases. Risks associated with HSCT can include graft-versus host disease, failure of the graft, and other toxicity related to the transplant. In one study of 31 people, the procedure was successful for 22 whose hemoglobin levels improved to the normal range, in seven the graft failed and they continued to live with thalassemia, and two died of transplantation-related causes.
Evolutionary advantage
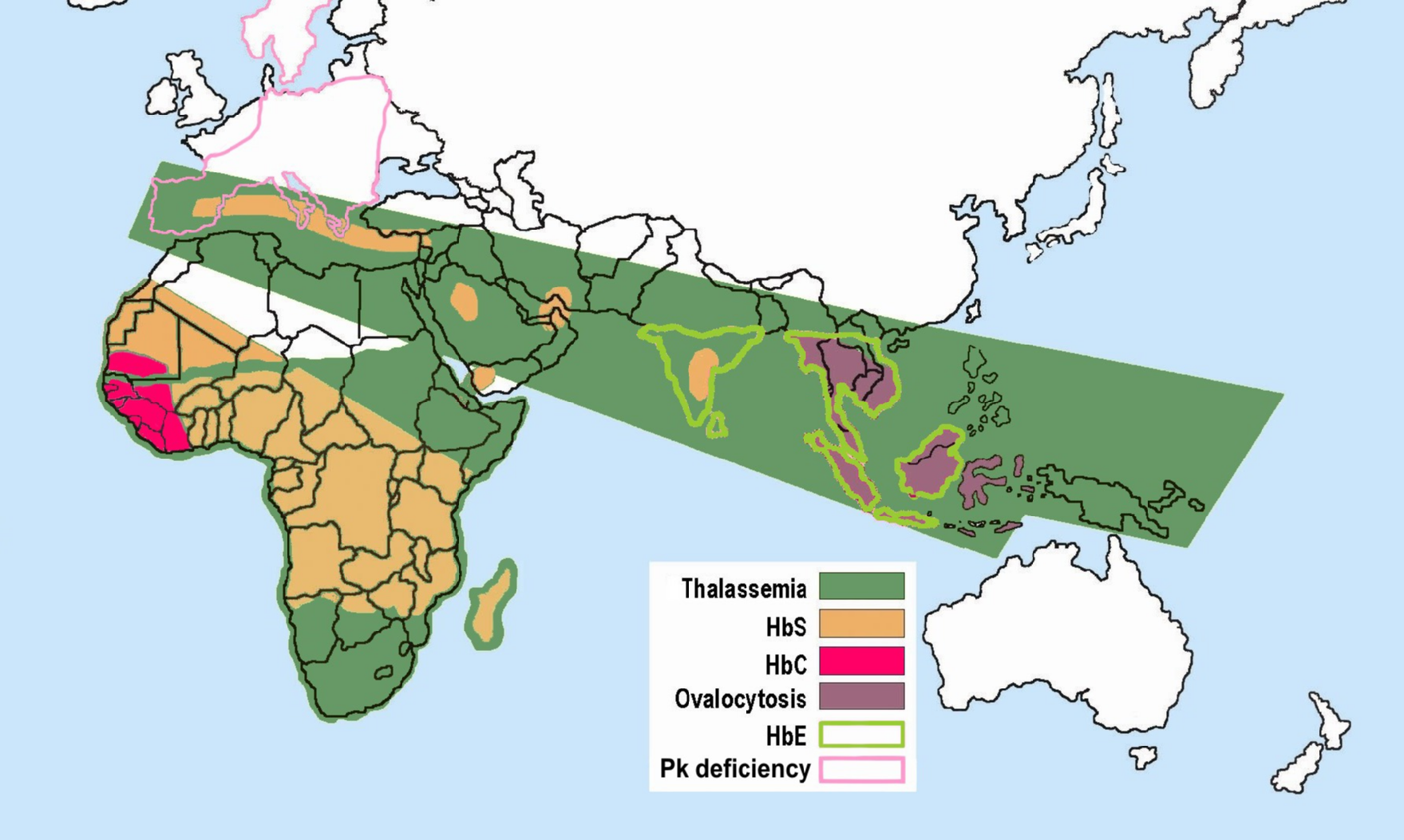
Main article: Malaria resistance
Some hemoglobinopathies seem to have given an evolutionary benefit, especially to heterozygotes, in areas where malaria is endemic. Having a mild form of alpha thalassemia has been demonstrated to protect against malaria and thus can be an advantage in malaria endemic areas, thus conferring a selective survival advantage on carriers (known as heterozygous advantage), and perpetuating the mutation.
Alpha thalassemia genes have a high prevalence in populations originating in sub-Saharan Africa, Mediterranean, Middle East, and southeast and east Asia; all areas which historically have been malaria endemic. The prevalence of these genes has increased in previously non-endemic areas as a consequence of migration flows, slave-trade, and colonization.
A number of mechanisms have been proposed to explain the increased chance of survival for the carrier of an abnormal hemoglobin trait.
Combination hemoglobinopathies
A combination hemoglobinopathy occurs when someone inherits two different abnormal hemoglobin genes. Alpha thalassemia can coexist with other hemoglobinopathies such as sickle cell disease and beta thalassemia. When alpha thalassemia carrier or trait combines with another hemoglobinopathy, the symptoms are generally those of the other .
References
References
- (1 January 1993). "Alpha-Thalassemia". GeneReviews.
- (December 2009). "BRS Pathology". [[Lippincott Williams & Wilkins]] medical.
- "Complications and Treatment {{!}} Thalassemia {{!}} Blood Disorders {{!}} NCBDDD {{!}} CDC".
- "Lanzkowsky's Manual Of Pediatric Hematology And Oncology 6th Edition ( 2016)".
- (27 January 2025). "Alpha Thalassemia".
- "Alpha-thalassemia - Symptoms, diagnosis and treatment {{!}} BMJ Best Practice".
- "Alpha thalassemia: MedlinePlus Genetics".
- (17 October 2022). "Thalassaemia - Thalassaemia carriers".
- (2023-11-10). "Clinical commissioning policy: allogeneic haematopoietic stem cell transplantation (Allo-HSCT) for adult transfusion dependent thalassaemia".
- Stuart H. Orkin. (2009). "Nathan and Oski's Hematology of Infancy and Childhood, Volume 1". Elsevier Health Sciences.
- (2010-05-28). "α-thalassaemia". Orphanet Journal of Rare Diseases.
- (May 2006). "The effect of alpha+-thalassaemia on the incidence of malaria and other diseases in children living on the coast of Kenya". PLOS Medicine.
- (2024-03-01). "Αlpha-thalassemia: A practical overview". Blood Reviews.
- (January 2005). "Acquired alpha-thalassemia in association with myelodysplastic syndrome and other hematologic malignancies". Blood.
- (17 Nov 2021). "Thalassemia-Thalassemia - Symptoms & causes".
- Pondarre, Corinne. (May 2021). "Orphanet: Hb Bart's hydrops fetalis".
- "Pathophysiology of alpha thalassemia".
- "Alpha Thalassemia in Children".
- (6 March 2022). "Thalassemias".
- (2017-10-24). "Thalassaemia - Symptoms".
- CDC. (2024-05-22). "About Thalassemia".
- (December 2017). "Impact of bone disease and pain in thalassemia". Hematology. American Society of Hematology. Education Program.
- (October 2016). "Craniofacial Characteristics of Thalassemia Major Patients". The Eurasian Journal of Medicine.
- (5 January 2025). "What Is Thalassemia?".
- (June 2022). "Non-transferrin bound iron". Clinica Chimica Acta; International Journal of Clinical Chemistry.
- (January 2016). "Endocrine Problems in Thalassemia".
- (25 Oct 2023). "Thalassaemia".
- (September 2022). "Thalassemia and the Spleen".
- (December 2009). "Infections in thalassemia and hemoglobinopathies: focus on therapy-related complications". Mediterranean Journal of Hematology and Infectious Diseases.
- (May 2019). "Hemoglobin H Disease".
- Pondarre, Corinne. (May 2021). "Hb Bart's hydrops fetalis".
- (March 2017). "An international registry of survivors with Hb Bart's hydrops fetalis syndrome". Blood.
- {{Emedicine. article. 958850. Pediatric Thalassemia. treatment
- (2025). "Thalassemia". StatPearls Publishing.
- (2018-01-25). "Treatment of Thalassemias".
- CDC. (2025-01-02). "Treatment of Thalassemia".
- Butler, Craig. (16 January 2025). "Transfusion Issues in Thalassemia".
- (17 October 2022). "Thalassaemia - Treatment".
- (2010). "Update on iron chelators in thalassemia". Hematology. American Society of Hematology. Education Program.
- (2023-05-09). "Treatment for osteoporosis in people with beta-thalassaemia". The Cochrane Database of Systematic Reviews.
- (17 Nov 2021). "Thalassemia-Thalassemia - Symptoms & causes".
- England, N. H. S.. (2023-11-10). "NHS England » Clinical commissioning policy: allogeneic haematopoietic stem cell transplantation (Allo-HSCT) for adult transfusion dependent thalassaemia".
- (2012-05-01). "Hematopoietic Stem Cell Transplantation in Thalassemia and Sickle Cell Anemia". Cold Spring Harbor Perspectives in Medicine.
- (2021-12-10). "Advances in the management of α-thalassemia major: reasons to be optimistic". Hematology.
- (2009). "Hematopietic Stem Cell Transplantation in Thalassemia and Related Disorders". Mediterranean Journal of Hematology and Infectious Diseases.
- Leigh, Suzanne. (2018-05-25). "Baby Born in World's First In Utero Stem Cell Transplant Trial {{!}} UC San Francisco".
- (2023-12-31). "A systematic review comparing allogeneic hematopoietic stem cell transplant to gene therapy in sickle cell disease". Hematology.
- (June 2011). "T cell-depleted hla-haploidentical stem cell transplantation in thalassemia young patients". Pediatric Reports.
- (May 2006). "The effect of alpha+-thalassaemia on the incidence of malaria and other diseases in children living on the coast of Kenya". PLOS Medicine.
- (December 2020). "Changing patterns in the epidemiology of β-thalassemia". European Journal of Haematology.
- (September 2012). "World distribution, population genetics, and health burden of the hemoglobinopathies". Cold Spring Harbor Perspectives in Medicine.
- (2013-05-16). "Hemoglobinopathies: Slicing the Gordian Knot of Plasmodium falciparum Malaria Pathogenesis". PLOS Pathogens.
- (2021-12-31). "Shared molecular basis, diagnosis, and co-inheritance of alpha and beta thalassemia". Blood Research.
- (2006). "Co-inheritance of α+-thalassaemia and sickle trait results in specific effects on haematological parameters". British Journal of Haematology.
This article was imported from Wikipedia and is available under the Creative Commons Attribution-ShareAlike 4.0 License. Content has been adapted to SurfDoc format. Original contributors can be found on the article history page.
Ask Mako anything about Alpha-thalassemia — get instant answers, deeper analysis, and related topics.
Research with MakoFree with your Surf account
Create a free account to save articles, ask Mako questions, and organize your research.
Sign up freeThis content may have been generated or modified by AI. CloudSurf Software LLC is not responsible for the accuracy, completeness, or reliability of AI-generated content. Always verify important information from primary sources.
Report